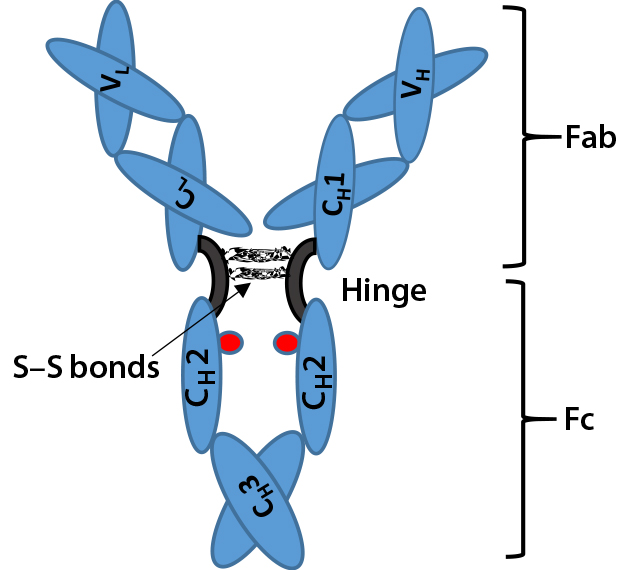
Figure 1: Immunoglobulin G (IgG) structure consists of four polypeptide chains, including two identical light chains (~25 kDa) and two identical heavy chains (~50 kDa). Each light chain consists of one constant domain (CL) and one variable domain (VL), and each heavy chain consists of three constant domains (CH1, CH2, and CH3) and one variable domain (VH). The Fab is the antigenbinding region containing hypervariable or complementarity-determining regions (CDRs), whereas the Fc region is highly conserved across molecular species. The hinge region contains two disulfide bonds and glycosylation (shown as red dots) at CH2 domain of Fc fragment.
Biosimilars are biologically derived pharmaceuticals intended to have clinical similarity to a legally marketed innovator product when that product’s patent or market exclusivity has expired. By contrast with generic small-molecule drugs, clinical performance of a biologic pharmaceutical is a function of its structural complexity and higher-order structure (HOS). Biomanufacturing controls of such complex products cannot fully ensure chemical similarity between an innovator product and putative biosimilar because minor differences in chemical modifications and HOS can significantly alter a product’s safety and efficacy.
Therefore, to substantiate claims of clinical functionality, a demonstration of bioequivalence is inadequate for biosimilar pharmaceuticals. This is different from regulatory approval for generic drugs, in which bioequivalence demonstration is adequate. The overall challenge in approving biosimilar pharmaceuticals is to enable scientific inference of similarity in safety and efficacy for a new biologically derived product compared with an innovator without repeating burdensome clinical studies.
In Part 1 of this two-part article, we focus on the scientific gaps that complicate determination of biosimilarity for one subset of biologically manufactured products: therapeutic monoclonal antibodies (TMAbs). Notably, many biosimilar products have been approved around the world, including in the United States: e.g., Sandoz’s Zarxio biosimilar to Amgen’s Neupogen (filgrastim) and Celltrion/Pfizer’s Inflectra biosimilar to Janssen/J&J’s Remicade (infliximab). First we briefly describe the regulatory landscape for the development of biosimilar TMAbs. Then we identify certain specific structural components of TMAbs — drug substances in Part 1, drug products in Part 2 — that warrant particular attention because alterations to them are likely to affect therapeutic safety and effectiveness. Finally, in Part 2 we consider whether studies of TMAb reference material can further the development of analytical industry standards to ensure comparability of putative biosimilar TMAbs with innovator TMAbs. And we suggest that the time is right to tie analytical industry standards and manufacturing controls to specific reductions in preclinical and clinical studies for regulatory approval of certain biosimilar TMAbs.
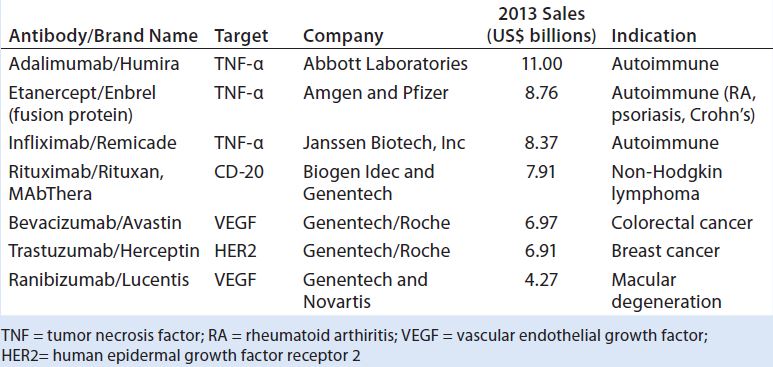
Table 1: Top-selling therapeutic monoclonal antibody (MAb) products in 2013
US Regulation of Biosimilar TMAbs
Biosimilars or “follow-on biologics” are potentially cost-effective alternatives to innovator biopharmaceuticals because they can significantly reduce the time it takes for product and process development with a lowered regulatory burden (1, 2). The FDA approved the first TMAb in 1986 (3), and about 30 TMAb products now account for annual sales in excess of US$60 billion (Table 1) (4). Pursuing the biosimilar path rather than using the original innovator product could save 15–30% or more in revenues (5–7).
Many innovator biologics will reach patent or market exclusivity expiry within the next few years (5, 6). In anticipation, US policy has been established to promote biosimilar approvals. Biosimilars are defined by the US law under Section 351(k) of the Public Health and Service Act (PHSA) as biologic products that are “highly similar to the reference product notwithstanding minor differences in clinically inactive components,” and without “clinically meaningful differences between the biological product and the reference product in terms of the safety, purity and potency of the product” (7).
Regulatory bodies had to consider scientific evaluation of comparability in pharmaceuticals long before biosimilars were considered as potential low-cost alternatives to innovator products (11, 12). The advent of biosimilars increases evaluation complexity. During innovator-product reviews such as for TMAbs, the issue of clinical comparability naturally arises because subtle manufacturing variations may yield glycosylation alterations or other posttranslational modifications to the TMAb product (13). Subtle molecular variations (microheterogeneities) can affect product potency and/or toxicity.
Fortunately, industry sponsors and regulatory reviewers already have experience in making scientific determinations from analytical and biological activity data. The International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) established guidelines in 2004 that describe a comparability exercise to ascertain whether a biologically manufactured product retained its quality despite changes in manufacturing (8, 9). The 1999 ICH Q6B guideline describes how to use analytical procedures to establish acceptance criteria for proteins made by cell culture expression systems (16). So despite manufacturing changes, an innovator TMAb can retain its clinical safety and efficacy previously demonstrated in clinical studies (10).
Guidelines established by ICH Q6B outline basic underlying principles used to set specifications, including characterization, analysis, and manufacturing controls (11, 16). Further, prespecified quantitative parameters — e.g., primary structure, glycosylation, disulfide structure, charge variants, size variants, biophysical characterization (secondary structure, tertiary structure, and thermal stability) ,and biological characterization — must be identified to determine acceptability of raw materials, excipients, and final products. Q6B also indicates the usefulness of pharmacopoeial tests for evaluating biologically manufactured pharmaceuticals in such areas as sterility, endotoxin levels, microbial limits, particulates, dose uniformity, and container volume. Release and shelf-life limits are established for potency and degradation products from both drug substance and finished drug product.
In 2006, the European Medicines Agency (EMA) published the first regulatory guidance for biosimilar approval (12, 13). Since then, many biosimilars have been approved in the European Union (14), one of which is the TMAb Inflectra (infliximab) that was also recently approved in the United States (15). The EMA has now published TMAb product-class–specific data for production, quality control, and nonclinical and clinical studies (16, 17). Those guidelines require demonstration of similarity — in terms of quality, safety, and efficacy — with a reference product authorized in the European Union. Although those products are not expected to be identical, even minor detected differences should be justified scientifically with respect to the safety and efficacy.
The World Health Organization (WHO) published analogous guidance in 2010 for “similar biotherapeutic products” (SBPs) through its Expert Committee on Biological Standardization (ECBS) (18). We suggest that experience and scientific evidence gained from biosimilar TMAbs already launched in Europe can provide additional insight to facilitate continuing biosimilar TMAb approval in the United States (19).
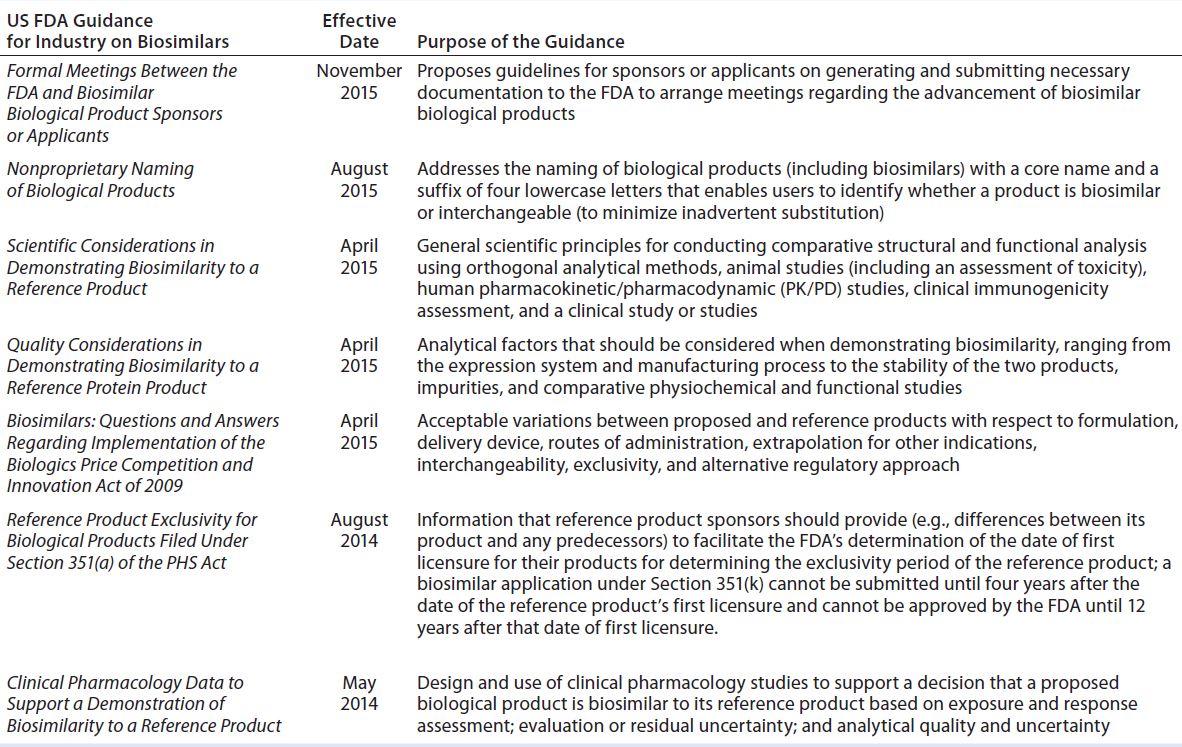
Table 2: US Food and Drug Administration (FDA) drafts and guidance documents on demonstrating biosimilarity
The 2009 US Biologics Price Competition and Innovation Act (BPCI) and the associated 2010 Patient Protection and Affordable Care Act (2010) incorporated a new regulatory pathway called 351(k) for biosimilar products (7). The US Food and Drug Administration (FDA) responded to those new laws by issuing draft guidance documents for industry sponsors and FDA reviewers (20–26) (Table 2). According to FDA guidelines for demonstrating biosimilarity, a biologics license application (BLA) will be evaluated and reviewed under provision 351(k) of the act for potential reduction of nonclinical and clinical studies (27). The agency decided on case-by-case evaluation of clinical data based on “totality of evidence” to approve biosimilars. It appears that the FDA does not accept that a standardized analytical comparison between an innovator and putative biosimilar could be applied across the entire class of TMAbs. Indeed, such a standard might be anticipated to complicate clinical studies initiated by sponsors.
In the sections that follow — here and in Part 2 — we describe product and process characteristics specific to TMAbs. We present knowledge gaps that limit the formation of industry- wide validation or standardization. Thus, we will show that we agree that the “case-by-case” nature of the current regulatory burden cannot be reduced at this time. Establishment of standards for cross-class product attributes are needed to reduce the regulatory burden in the future.
Drug Substance: Conserving Primary Structure
Most TMAbs are of the IgG-class of immunoglobulins. IgGs are ~150 kDa in size, composed of four polypeptide chains, including two identical light chains (~25 kDa) and two heavy chains (~50 kDa) (Figure 1). The four constituent polypeptides in the antibody interact to form a three-dimensional (3D) “Y” shape. That 3D structure confers physiological specificity. The Fab moiety binds antigens and contains the complementarity-determining region, whereas the Fc moiety is highly conserved.
Protein structure can be described as primary (linear amino-acid sequence), secondary (sectional folding of alpha helixes and beta sheets), tertiary (3D molecular shape), and quaternary (subunit domains assembled into single entity). Modification to the primary polypeptides, such as those following translation of the amino-acid sequence (e.g., glycosylation, deamidation, and degradation), can affect product safety and efficacy (28). Among the most noted posttranslational modifications (PTMs) for TMAbs are their varied N-linked glycan structures, which include galactosylation, fucosylation, mannosylation, and sialylation.
Glycosylation is a highly variable and heterogeneous process that depends on such factors as clonal variation, production cell line, media, and culture conditions (29, 30). We note, however, that some changes in glycosylation stemming from manufacturing changes (e.g., the pre- to postchange scenario described above) have been accepted by FDA, allowing for heterogeneity in glycan structures to be accommodated (31–33). In such cases, manufacturing process details were made available. That information probably would not be available to a biosimilar TMAb sponsor intending to compete with an innovator.
Below, we offer examples in which glycosylation of specific regions or portions of TMAbs could be critical for an antibody’s pharmacologic function. Thus, differences from an innovator’s manufacturing controls when a competitor attempts to create a biosimilar TMAb can be expected to have ramifications for therapeutic safety and efficacy.
Fc Region of TMAbs
Example 1: N-linked glycosylation is conserved in humans at Asn-297 of the CH2 domain on the Fc portion of IgG (IgG-Fc), below the hinge region. The IgG-Fc portion binds to Fcγ receptors (FcγRs) on the surface of effector immune cells, resulting in antibody-dependent, cell-mediated cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), phagocytosis, oxidative burst, and secretion of inflammatory mediators (34). Glycosylation of the IgG-Fc has been shown to be critical for mediating the binding interaction with FcγRs to trigger ADCC. In particular, the lower hinge region of IgG-Fc affects binding and activation of FcγRIII (34, 35).
Example 2: Core fucosylation (α 1, 6-fucose linked to N-acetylglucosamine) reduces binding affinity to the FcγRIIIa receptor and thus degrades ADCC (36–38). Similarly, nonfucosylated antibodies evade the inhibitory effects of serum IgG on ADCC through its high binding affinity to FcγRIIIa expressed on natural killer (NK) cells (39). Alternatively, glycosylation at Asn-162 of the FcγRIIIa receptor is critical for binding to the Fc region (40). For example, MAbs produced by Chinese hamster ovary (CHO) cells typically have complex biatennary structures with little bisecting N-acetylglucosamine (GlcNAc) and a high level of core fucosylation. Overexpression of N-acetylglucosaminetransferase III in such cell lines increases bisecting GlcNAc and nonfucosylated oligosaccharides on MAbs and thus raises ADCC (41).
Example 3: Glycosylation of the Fc region is also indispensable for maintaining a long catabolic half-life in vivo (42). IgGs containing high-mannose glycans have shown increased serum clearance (43) and addition of terminal sialic acid to the Fc glycan leads to upregulation of surface expression of the FcγRIIb on inflammatory cells, thereby initiating an antiinflammatory cascade (44, 45). In addition, the presence of N-glycolylneuraminic acid (NeuGC or NGNA) in recombinant therapeutic glycoproteins expressed in nonhuman cell cultures may be immunogenic and potentially relevant to half-life, efficacy, and adverse events (46, 47). Also, glycosylation at the CH2 domain of IgG1-Fc affects C1q/C1 binding and activation leading to inflammatory and immunoregulatory responses consistent with CDC (48).
Thus, an abundance of scientific evidence indicates that specific glycosylation of the Fc region can be quantitatively evaluated (49). Fc glycoprofiles are expected to be strongly associated with safety and efficacy.
Fab Region of TMAbs
Example 4: The oligosaccharide galactose-α-1,3-galactose on the Fab portion of the heavy chain of cetuximab, a chimeric IgG1 MAb against the epidermal growth factor receptor (EGFR), is linked to unwanted immunological response (50). It is also notable that 15–25% of the IgG-Fab variable domain contains N-linked oligosaccharides occupied by biantennary carbohydrates with variable sialic acid content (51, 52). Variable-domain glycosylation on Ab function suggests that physiological performance may be comparably varied (52), including a potential relationship to Ab half-life (53).
To achieve biosimilarity of TMAbs, their primary structure (including posttranslational modifications) must be rigorously conserved. Whereas ample data are available relating the physiologic and clinical function of TMAbs to their posttranslational chemistry, the body of science is not comprehensive. So this information cannot by itself predict clinical outcomes.
Biosimilar developers face an even more daunting manufacturing challenge than innovators do when controlling primary structure of a T-MAb. Unlike innovator companies, developers of biosimilar TMAbs have no access to the original, proprietary manufacturing process data from the originator. Therefore, biosimilar developers must completely reverse-engineer their manufacturing methods and process controls using publicly available scientific knowledge about the innovator TMAbs (32). In addition, even with a comparable product from a reverse-engineered manufacturing process, biosimilar TMAbs developers must attempt to recreate a new, therapeutically equivalent product formulation (see part 2) — whereas innovators simply maintain quality control over an approved TMAb production process and formulation.
Drug Substance Biosimilarity Confirmation
Analysis of TMAb higher-order structure (HOS) is just as critical as primary structure when developers want to establish biosimilarity. Protein misfolding, for example, can have an impact on drug safety by eliciting unwanted immune responses or other off-target physical responses. A number of biophysical techniques are used to characterize TMAb HOS: e.g., X-ray crystallography, nuclear magnetic resonance (NMR) imaging, Fourier-transform infrared (FTIR) spectroscopy, circular dichroism (CD), differential scanning calorimetry (DSC), and isothermal calorimetry. Because no single analytical assay is sufficient to determine comparability between a biosimilar and originator product, developers use state-of-the-art orthogonal analytical techniques (11, 54). In some cases, surrogate bioassays can be used to confirm the absence of changes in HOS introduced by the new manufacturing process of biosimilars. Changes in such characteristics can affect product potency (20, 21).
For biosimilars, a product’s biological activity also can be an indicator of stability and manufacturing process consistency across batches. In general, however, bioassay variability precludes the sensitivity of such assays to detect minor changes in potency. So although they are helpful, biological and/or functional assays may not fill a gap in analytical assay sensitivity to detect minor conformational differences between biosimilar TMAbs and innovator products. It is important to note that no analytical test or combination for HOS has yet been sufficiently validated for analytical testing as a substitute for clinical studies in the development of a biosimilar TMAbs drug substance. Without such validation, creating a quantitative industry standard to establish HOS similarity seems premature. Instead, renewed investment in developing predictable and standardized HOS measurements of validated reference materials is envisioned as a path forward.
Looking Ahead
Next month, we will conclude this two-part discussion by examining drug product issues (especially related to aggregation and excipients) and considering the possibility of calibrating analytical methods against validated reference antibodies. We also will present a case study before offering some conclusions and perspectives.
Acknowledgments
This work was supported with funds and a fellowship award from the Global Biological Standards Institute (GBSI). We thank Dr. Anthony Mire-Sluis at AstraZeneca (California) and Dr. Hans Ebbers at the Utrecht Institute for Pharmaceutical Sciences (The Netherland) for their feedback and critical review. We are grateful to University of Maryland and GBSI staff for their support of this project. In particular, we thank Mark Gibson from GBSI for helping to put together early drafts of this work. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agency.
References
1 Ahmed I, Kaspar B, Sharma U. Biosimilars: Impact of Biologic Product Life Cycle and European Experience on the Regulatory Trajectory in the United States. Clin. Ther. 34(2) 2012: 400–419.
2 Gitter DM. Informed By the European Union Experience: What the United States Can Anticipate and Learn from the European Union’s Regulatory Approach to Biosimilars. Seton Hall Law Rev. 41(2) 2011: 559–592.
3 Smith SL. Ten Years of Orthoclone OKT3 (Muromonab-CD3): A Review. J. Transpl. Coord. 6(3) 1996: 109–119; quiz 120–121.
4 Aggarwal RS. What’s Fueling the Biotech Engine: 2012 to 2013. Nat. Biotechnol. 32(1) 2014: 32–39.
5 Grabowski H. Follow-On Biologics: Data Exclusivity and the Balance Between Innovation and Competition. Nat. Rev. Drug Discov. 7(6) 2008: 479–488.
6 Lanthier M, Behrman R, Nardinelli C. Economic Issues with Follow-On Protein Products. Nat. Rev. Drug Discov. 7(9) 2008: 733–737.
7 Public Law 111-148, Section 1695. Biologics Price Competition and Innovation Act. Provisions of the Patient Protection and Affordable Care Act. 110th Congress: 23 March 2010.
8 ICH Q5E. Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process. US Fed. Reg. 70(125) 2005: 37861–37862.
9 Chirino AJ, Mire-Sluis A. Characterizing Biological Products and Assessing Comparability Following Manufacturing Changes. Nat. Biotechnol. 22(11) 2004: 1383–1391.
10 Comment (Docket FDA-2011-D-0618). Draft Guidances Relating to the Development of Biosimilar Products: Public Hearing; Request for Comments. Novartis AG: Basel, Switzerland, 2012.
11 ICH Q6B. Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products. US Fed. Reg. 64, 1999: 44928.
12 EMA/CHMP/BMWP/42832. Guideline on Similar Biological Medicinal Products Containing Biotechnology Derived Proteins As Active Substance: Non-Clinical and Clinical Issues. European Medicines Agency: London, UK, 2006.
13 CHMP/BMWP/49348. Guideline on Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins As Active Substance: Quality Issues. European Medicines Agency: London, UK, 2006.
14 Tsiftsoglou AS, Ruiz S, Schneider CK. Development and Regulation of Biosimilars: Current Status and Future Challenges. BioDrugs 27(3) 2013: 203–211.
15 Chowdhury BA. BLA Approval 125544. United States Food and Drug Administration: Rockville, MD, April 2016.
16 EMEA/CHMP/BWP/157653/2007. Guideline on Production and Quality Control of Monoclonal Antibodies and Related Substances. European Medicines Agency: London, UK, 2009.
17 EMA/CHMP/BMWP/403543/2010. Guideline on Similar Biological Medicinal Products Containing Monoclonal Antibodies: NonClinical and Clinical Issues. European Medicines Agency: London, UK, 2012.
18 Guidelines on Evaluation of Similar Biotherapeutic Products (SBPs). World Health Organisation: Geneva, Switzerland, 2010.
19 Schneider CK, et al. Setting the Stage for Biosimilar Monoclonal Antibodies. Nat. Biotechnol. 30(12) 2012: 1179–1185.
20 CDER/CBER. Guidance for Industry: Quality Considerations in Demonstrating Biosimilarity of a Therapeutic Protein Product to a Reference Product. US Food and Drug Administration: Rockville, MD, April 2015.
21 CDER/CBER. Guidance for Industry: Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. US Food and Drug Administration: Rockville, MD, April 2015.
22 CDER/CBER. Guidance for Industry: Biosimilars — Questions and Answers Regarding Implementation of the Biologics Price Competition and Innovation Act of 2009. US Food and Drug Administration: Rockville, MD, April 2015.
23 CDER/CBER. Guidance for Industry: Clinical Pharmacology Data to Support a Demonstration of Biosimilarity to a Reference Product. US Food and Drug Administration: Rockville, MD, May 2014.
24 CDER/CBER. Guidance for Industry: Formal Meetings Between the FDA and Biosimilar Biological Product Sponsors or Applicants. US Food and Drug Administration: Rockville, MD, November 2015.
25 CDER/CBER. Draft Guidance for Industry: Nonproprietary Naming of Biological Products. US Food and Drug Administration: Rockville, MD, August 2015.
26 CDER/CBER. Draft Guidance for Industry: Reference Product Exclusivity for Biological Products Filed Under Section 351(a) of the PHS Act. US Food and Drug Administration: Rockville, MD, August 2014.
27 Wang J, Chow SC. On the Regulatory Approval Pathway of Biosimilar Products. Pharmaceuticals 5(4) 2012: 353–368.
28 Walsh G, Jefferis R. Post-Translational modifications in the Context of Therapeutic Proteins. Nat. Biotechnol. 24(10) 2006: 1241– 1252.
29 Patel TP, et al. Different Culture Methods Lead to Differences in Glycosylation of a Murine IgG Monoclonal Antibody. Biochem. J. 285(Pt 3) 1992: 839–845.
30 Schmelzer AE, Miller WM. Hyperosmotic Stress and Elevated pCO2 Alter Monoclonal Antibody Charge Distribution and Monosaccharide Content. Biotechnol. Prog. 18(2) 2002: 346–353.
31 McCamish M, Woollett G. Worldwide Experience with Biosimilar Development. MAbs 3(2) 2011: 209–217.
32 McCamish M, Woollett G. The State of the Art in the Development of Biosimilars. Clin. Pharmacol. Ther. 91(3) 2012: 405–417.
33 Schiestl M, et al. Acceptable Changes in Quality Attributes of Glycosylated Biopharmaceuticals. Nat. Biotechnol. 29(4) 2011: 310–312.
34 Hulett MD, Hogarth PM. Molecular Basis of Fc Receptor Function. Adv. Immunol. 57, 1994: 1–127.
35 Radaev S, Sun P. Recognition of Immunoglobulins By Fc Gamma Receptors. Mol. Immunol. 38(14) 2002: 1073–1083.
36 Matsumiya S, et al. Structural Comparison of Fucosylated and Nonfucosylated Fc Fragments of Human Immunoglobulin G1. J. Mol. Biol. 368(3) 2007: 767–779.
37 Ferrara C, et al. Unique Carbohydrate-Carbohydrate Interactions Are Required for High Affinity Binding Between FcgammaRIII and Antibodies Lacking Core Fucose. Proc. Natl. Acad. Sci. USA 108(31) 2011: 12669–12674.
38 Shields RL, et al. Lack of Fucose on Human IgG1 N-Linked Oligosaccharide Improves Binding to Human Fcgamma RIII and Antibody-Dependent Cellular Toxicity. J. Biol. Chem. 277(30) 2002: 26733–26740.
39 Iida S, et al. Nonfucosylated Therapeutic IgG1 Antibody Can Evade the Inhibitory Effect of Serum Immunoglobulin G on Antibody-Dependent Cellular Cytotoxicity Through Its High Binding to FcgammaRIIIa. Clin. Cancer Res. 12(9) 2006: 2879–2887.
40 Zeck A, et al. Cell Type-Specific and Site Directed N-Glycosylation Pattern of FcgammaRIIIa. J. Proteome Res. 10(7) 2011: 3031–3039.
41 Yamane-Ohnuki N, Satoh M. Production of Therapeutic Antibodies with Controlled Fucosylation. MAbs 1(3) 2009: 230–236.
42 Jones AJ, et al. Selective Clearance of Glycoforms of a Complex Glycoprotein Pharmaceutical Caused By Terminal N-Acetylglucosamine Is Similar in Humans and Cynomolgus Monkeys. Glycobiol. 17(5) 2007: 529–540.
43 Goetze AM, et al. High-Mannose Glycans on the Fc Region of Therapeutic IgG Antibodies Increase Serum Clearance in Humans. Glycobiol. 21(7) 2011: 949–059.
44 Anthony RM, Ravetch JV. A Novel Role for the IgG Fc Glycan: The Anti-Inflammatory Activity of Sialylated IgG Fcs. J. Clin. Immunol. 30 (Sup 1), 2010: S9–S14.
45 Kaneko Y. Anti-Inflammatory Activity of Immunoglobulin G Resulting from Fc Sialylation. Science 313(5787) 2006: 670–673.
46 Ghaderi D, et al. Implications of the Presence of N-Glycolylneuraminic Acid in Recombinant Therapeutic Glycoproteins. Nat. Biotechnol. 28(8) 2010: 863–837.
47 Yu C, et al. At Least Two Fc Neu5Gc Residues of Monoclonal Antibodies Are Required for Binding to Anti-Neu5Gc Antibody. Sci. Rep. 7, 2016: 20029.
48 Jefferis R. Isotype and Glycoform Selection for Antibody Therapeutics. Arch. Biochem. Biophys. 526(2) 2012: 159–166.
49 Flynn GC, et al. Naturally Occurring Glycan Forms of Human Immunoglobulins G1 and G2. Mol. Immunol. 47(11–12) 2010: 2074–2082.
50 Chung CH, et al. Cetuximab-Induced Anaphylaxis and IgE Specific for Galactose-Alpha-1,3-Galactose. N. Engl. J. Med. 358(11) 2008: 1109–1117.
51 Spiegelberg HL, et al. Localization of the Carbohydrate Within the Variable Region of Light and Heavy Chains of Human Gamma G Myeloma Proteins. Biochemistry 9(21) 1970: 4217–4223.
52 Leibiger H, et al. Variable Domain-Linked Oligosaccharides of a Human Monoclonal IgG: Structure and Influence on Antigen Binding. Biochem. J. 338(Pt 2) 1999: 529–538.
53 Huang L, et al. Impact of Variable Domain Glycosylation on Antibody Clearance: An LC/MS Characterization. Anal. Biochem. 349(2) 2006: 197–207.
54 Berkowitz SA, et al. Analytical Tools for Characterizing Biopharmaceuticals and the Implications for Biosimilars. Nat. Rev. Drug Discov. 11(7) 2012: 527–540.
Simran J. Kaur is an ORISE research fellow at the US Food and Drug Administration; Darryl Sampey is president and CEO of BioFactura, Inc. (Frederick MD); Lester W. Schultheis is director of the Maryland Regulatory Science Initiative, part of the Fischell Department of Bioengineering at the University of Maryland (College Park); and Leonard P. Freedman is president of the Global Biological Standards Institute (Washington, DC). Corresponding author William E. Bentley is co–principal-investigator of the University of Maryland’s Center of Excellence in Regulatory Science and Innovation (M-CERSI) and distinguished professor of the University of Maryland, 3122 Kim Engineering Building, Fischell Department of Bioengineering, University of Maryland, College Park, 20742; 1-301-405-4321, fax 1-301-405-9953; bentley@umd.edu.