No consensus concerning biosimilar-related terminology and definitions has yet been achieved (1,2,3). Biosimilars may be defined as biopharmaceuticals slated for generic-drug–like, abbreviated, comparisons-based approvals through a formal biosimilar approval pathway in the United States, European Union, and/or other highly regulated and developed countries based on a demonstration of substantial (bio)similarity to a reference product.
As required in the United States, biosimilar active agents (those involving recombinant proteins) must be identical in primary sequence with their reference products. Analytical and comparative bioequivalence/pharmacokinetic (PK) clinical testing must support a lack of significant differences (particularly in efficacy and safety) between the biosimilar and the reference product. Biosimilars is also sometimes used to refer to biogenerics, including the ∼150 mostly lower-end “knock-offs” of established major-market innovator products not made to good manufacturing practice (GMP) standards and sold in lesser-regulated international markets.
Biobetters refers to rather (bio)similar follow-on biopharmaceuticals that are too dissimilar (based on analytical, preclinical, and/or clinical testing) to their reference products to receive biosimilar approval. They receive conventional full market approval. Biobetters generally involve the “same” or similar active agent, either in a different physical form (e.g., different formulation or slow-release complex) or as a chemical substructure (e.g., PEGylated or expressed as a fusion protein).
The European Union is years ahead of the United States, having approved its first biosimilar in 2006 with over a dozen biosimilar filings approved since then. Uptake has been slow or negligible in nearly all European countries, however, with the total market for all EU biosimilars around US$400–$500 million at most. That equates to an average of about $40 million/year market for each product, an inadequate market by most pharmaceutical (and particularly, biopharmaceutical) standards. About $400 million is more typical for a single mainstream biopharmaceutical product.
Biosimilar markets in other countries — e.g., Canada and Japan — also are small parts of the big picture. Overall, biosimilars have yet to make a real impact on the biopharmaceutical market. With the United States being the primary market for innovative pharmaceuticals and US payers fully expected to force rapid, widespread adoption of less-expensive biosimilar products, the country will be the most important market for biosimilars by far.
The Biotechnology Information Institute (BII) began an effort late in 2011 to develop the most comprehensive pipeline database for biosimilars and biobetters. FirstWord Pharma published a print-format version of it in September 2012 (5). And later this year, BII will launch a more extensive online database (6).
This project involves two major tasks:
-
projecting patent and other exclusivity expirations for all potential US and EU reference products — completed for 119 potential reference products so far, nearly all recombinant proteins, with this information now integrated into the BIOPHARMA (reference products) database (7)
-
continuous compiling all biosimilars and biobetters reported in development along with related companies’ involvement from all nonproprietary sources.
Target Reference Products
Much as with generic drugs, essentially every currently successful biopharmaceutical is or will be a potential target for biosimilars development. Currently more than 425 biopharmaceutical products are approved in the United States and/or European Union, including more than 140 recombinant proteins approved in the former with total annual worldwide sales over $100 billion (7). The primary targets for US biosimilar development will be those recombinant proteins, including monoclonal antibodies (MAbs), and a few high-purity protein products. So most biosimilar targets are current US-marketed recombinant therapeutics, particularly those with sufficiently large markets to make even a small percentage profitable for competing biosimilars (and biobetters).
Obviously, the prime reference product targets are those enjoying the highest sales. Currently 40 recombinant proteins have blockbuster (≥$1 billion/year) markets, and another 18 have sales between $500 million and $1 billion. Even attaining just a 5–10% market share with a biosimilar version of a blockbuster (a market of $50–100 million/year) is an attractive proposition, particularly considering that the costs for biosimilar development of biosimilars should only rarely exceed that level. Such costs are generally projected to be about $25–60 million (but can be much higher for some products, such as when purchase of the reference product for trials could cost tens of millions alone).
Herein I concentrate primarily on biosimilars, but biobetters also will be major competitors taking market share from originator products. They will be just as important as biosimilars and new me-too/same-indication innovator products as future competitors on the market. When Can Biosimilars Enter the US Market?
Biosimilars cannot be marketed until all relevant government-granted exclusivities have expired for their associated reference products:
-
granted patents, multiple and diverse types of which protect nearly every reference product to varying extents to effectively provide market exclusivity (Other factors, such as patent extensions, can make patent-based market exclusivity last longer.)
-
market exclusivity granted by regulatory agencies, primarily related to orphan-drug status (seven years in the United States, 10 in the European Union)
-
data exclusivity granted by regulatory agencies, with the US biosimilars legislation giving reference products (those with full BLAs) 12 years of data exclusivity after full approval, during which the US FDA will not approve a biosimilar based on a given reference product (the European Medicines Agency grants 10 years of such protection).
BII estimated US and EU biosimilar and biobetter launchability dates for 119 candidate reference products, nearly all of them recombinant but including a few nonrecombinant proteins (not vaccines and blood products). The launchability date for a biosimilar is the latest expiration date for relevant patents as well as data and market exclusivity. Because biobetters involve full approvals (and thus are not affected by data exclusivity), their launchability dates would be simply the latest date of patent expiration or market exclusivity granted.
The length of data exclusivity for reference/innovator products was a major political issue during US debates about biosimilars legislation. Data exclusivity is considered a means of incentivizing product innovation by ensuring that product developers will have sufficient (minimal) market monopoly to assure adequate payback/profits on their investments. This issue will not go away, as many vested interests and politicians wanting to save money through biosimilars competition continue to revive proposals to shorten the current 12-year data exclusivity to seven years (e.g., as in the Obama administration’s 2014 FY budget proposed in April 2013).
How important or relevant will data exclusivity be to providing exclusivity for reference/innovative biologics in the United States?Table 1 compares projected US patent expiration dates to market and data exclusivity dates. (Determining relevant patents is a difficult and subjective task, and biosimilar developers need to perform more in-depth analyses of specific products than was possible in this study involving 119 of them.) With both US and EU patents, no type of exclusivity granted by regulatory agencies governs biosimilar launchability for 86–87% of the 119 current reference products BII evaluated. Thus, patents are almost always the determinant of biosimilar launchability in both regions.
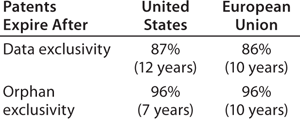
The average period of US reference-product patent expirations after a product’s original approval is 15.8 years. The range was –4 years (negative years for three products that received approval after their patent expirations) to an estimated 34 years patent coverage (to 2020) from the original approval for interferon alfa-2b (Merck & Company’s Intron A approved in 1986). That’s even greater than the 30 years for etanercept (Amgen’s Enbrel), which recently received a new patent extending its coverage to 2028. US new chemical entity (NCE)–based five-year market exclusivity granted to novel active agents is irrelevant in this analysis, with patents exceeding it 100% of the time.
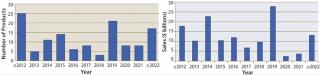
When will biosimilars enter the US market?Figure 1 estimates the number of reference products by their year of US launchability (generally patent expiration), and Figure 2 shows the total cumulative worldwide sales for those products by year. Over $100 billion of current reference/innovator products will become susceptible to biosimilar competition by 2020. A good number of biosimilars should enter the market in the next few years, with another large group (including MAb blockbusters) becoming launchable (patents expiring) towards the end of the decade. Among the 119 marketed recombinant proteins/MAbs evaluated by BII, about 30 are susceptible to biosimilar competition already. However, most of those involve some complications (e.g., high-tech delivery devices or small markets) such that they have yet to be targeted for biosimilar application filings.
How Many Products Are in the Pipeline?
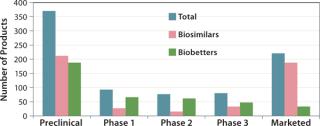
The BII pipeline database currently includes 514 candidate biosimilars and 402 biobetters, a total of 916 products in development concerning 119 of the >140 recombinant proteins that are currently approved in the United States. Compare that number with the 907 products the Pharmaceutical Research and Manufacturers’ Association (PhRMA) reports for the US biopharmaceutical (originators) clinical development pipeline (8).
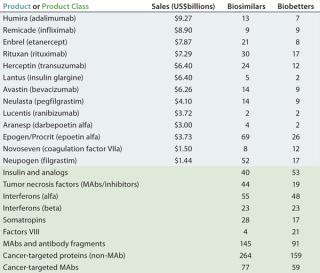
Table 2 lists some current biosimilars and biobetters in development according to their reference products (with recent annual sales) and product class. As with most pharmaceutical pipelines, the majority of products represented are in preclinical stages; as is normal, most will fail or be abandoned along the way. We can assume higher trial entry and approval rates for biosimilars than for innovative products, but the truth remains to be seen.
Candidate products have yet to be disclosed from many likely major biosimilar players, including some of the largest international pharmaceutical and generic drug companies, so they are not represented in the BII database. A number of those companies probably will be licensing-in products that are now being developed by other (smaller), companies. Otherwise, the BII analyses include every company known to be active in developing biosimilars and/or biobetters.
Where are biosimilars in the pipeline? Very few biosimilars in development have yet entered clinical trials to support US, EU, or other highly regulated country approvals. Almost half of the 514 biosimilar candidates (232) are in preclinical stages. With the exception of EU-approved products, most “marketed” biosimilars are essentially biogenerics in minimally regulated international commerce (needing new trials for major-market approval). Thus, you can consider ≥80% of marketed biosimilars to be at the preclinical stage in terms of US and EU markets. Relatively few biosimilars are in clinical trials overall, particularly compared with biobetters, which can be thought of as nothing new, simply business as usual (partially innovative, next-generation, me-too products).
So far, biosimilar development has been orderly in general, with no desperation or companies racing to be first into trials or to file for approvals. Few developers are rushing to get trials done early for major targets. A few large companies that took early leads in doing so (before any useful FDA guidance) have recently suspended their efforts. For example, Celltrion, Teva, and Samsung have halted trials of biosimilar rituximab (Rituxan from Genentech and Biogen Idec).
Unlike generic drugs, for which the first US approval receives six months of market exclusivity, biosimilars get no reward for being first to market. In fact, the first companies to file will probably bear the brunt of resolving patent disputes, which could cost tens of millions of dollars, allowing products filed/approved later to avoid much of that trouble. The first to file also will probably have to face more regulatory hurdles. An unknown number of companies may be holding off or proceeding slowly as they look forward to future interchangeable approvals for major market countries — or at least for better guidance from the FDA. With most of the best reference targets coming off patent in coming years, developers still have time to organize trials for the most target-worthy ones.
A large number of biosimilars will enter trials in the next few years. Such studies can be completed relatively quickly because they will usually be comparative bioequivalence trials only rather than full phase 3–type trials for proving efficacy and safety. With some reference products, however, biosimilar trials will be expensive and complex, including treatments of cancer, arthritis, and other complex, chronic diseases for which purchasing reference products for comparison alone can easily cost tens of millions of dollars. Just purchasing those reference products for testing can be a challenge. Some are simply not available (that is, sold only by their manufacturers, which have no obligation to sell products to potential competitors). It may be difficult for some developers to find trial sites and specialists that are not already under contract or don’t have conflicts of interest from working with reference-product companies. The ability to conduct trials probably will be the main choke-point in biosimilar development. Those companies that do not reserve contract research organizations (CROs) and trial sites ahead of the pack may encounter problems later on. Who’s Developing Biosimilars?
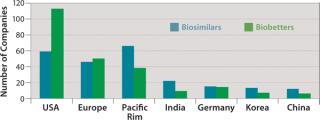
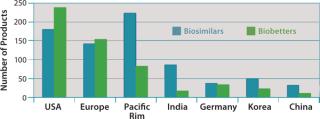
Figure 3 shows the geographic distribution of companies involved in development of biosimilars and biobetters, and Figure 4 shows the geographic distribution of the associated products in development. Asia and the Pacific Rim collectively have the most biosimilar companies and products in development, followed by the United States and Europe. It remains to be seen how many Asian and other foreign products will ultimately enter US-oriented clinical trials. Far ahead in innovative biopharmaceutical development, the United States is also the clear leader in terms of developing incrementally innovative biobetter products.
Individual companies in different regions have different numbers of products in development. On average, Asian companies claim roughly about twice as many biosimilars in development compared with their US counterparts, which in turn have about twice as many biobetters in development. What Asian companies report as a product in development, however, may be different in the level of effort involved than those based in the United States and elsewhere. Table 3 lists companies with the most biosimilar products (≥10) reported in development.
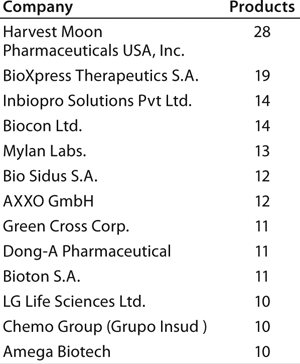
With by far the largest portfolio, Harvest Moon Pharmaceuticals USA Inc. (one of the very first biosimilar developers) is an outlier, and it is so far the only US company with ≥10 biosimilars reported in development. Sooner or later, however, many of the largest US and European companies will be disclosing their development portfolios, including the most likely future major players (particularly in terms of marketing) with ≥10 candidates.
Figure 5 indicates the size of companies that have reported biosimilars (160 total) or biobetters (173 total) in development as well as the total number of companies involved (including those not yet disclosing their pipeline products). The distinct lack of established, public, and midsized US biotechnology companies among biosimilar developers is not apparent in the figure. But those companies are generally preoccupied with innovative product development and biobetters.
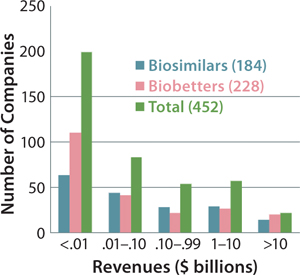
Figure 5:
How Will the US Market Evolve?
With few biosimilars in trials so far, it is too early to attempt to predict the likely size and diversity of the US market. However, it is becoming clear that a large number of biosimilars (and biobetters) will enter the market as their reference product patents expire. With many new entrants to the industry, bioprocessing equipment and services sales have increased already. Contract manufacturing organizations (CMOs) report an overall 15% increase in business from biosimilars. Major early milestones (and barriers) for all biosimilar development programs will include reference-product testing (easily costing >$1 million) using such methods as mass spectrometry; bioprocess development (with CMOs commonly quoting ∼$3 million to get to hundreds of liters in full CGMP compliance); and clinical trials (minimally costing millions of dollars and the most expensive effort).
Biosimilars will lead to biopharmaceutical revenue contraction, at least for the competing products involved. Because of increased competition as products come off patent and follow-ons enter the market, the market size will contract for each reference product and its biosimilars, biobetters, and other me-too products as they all compete, presumably at lower prices. Therefore, although biosimilars are very much expanding the market in numbers of players and products, the overall size of the markets for each group of affected products will contract as less-expensive versions gain traction.
BII believes that the US biosimilar market will be more like that of generic rather than innovator drugs. Much as the blockbuster asthma/allergy drug Singulair (montelukast sodium) recently went generic with 10 products approved by the FDA on the same day, some 10 or more biosimilars could rapidly enter the market for each successful reference product. Applying that to the 40 reference products with current ≥$1 billion/year sales suggests ≥400 biosimilars entering the market.Presuming that just five biosimilars enter the market for the 18 products with sales ≥$500 million, that’s an additional 90 products — for a total of ∼500 follow-on biopharmaceuticals entering the US market in this decade alone!
And that is probably a low-ball estimate, with more products and players potentially involved. If for each major reference product, for example, biosimilars are launched by just three Big Pharma companies, three smaller biotech companies, three of the largest generic drug companies, and three foreign new entrants to the US market, then that equates to a dozen biosimilars for one reference product. Those numbers are likely to be higher in reality, with most of the largest Big Pharma and generic companies expected to add biosimilars to round out their portfolios sooner or later. In other words, there will be a large amount of competition.
What types of companies will make up the US biosimilars market? All types and sizes from everywhere will be represented, but most of those involved will tend to be either quite large or quite small. Will too many players make for too much competition? Maybe so — especially for those companies expecting high, innovator-like, profit margins for biosimilars.
What will pricing models look like? The current consensus is based on limited EU experience and projects biosimilars entering the US market to be discounted 20–30% relative to their reference products. But BII believes that is too low an estimate for a market where insurers/payers will rapidly switch to biosimilar use, forcing companies into price competition (as with generic drugs). Already a future major player, Samsung Bioepis (the joint venture of Samsung with Biogen Idec) has reported that it intends to launch a full portfolio of biosimilars — including MAbs — in the United States at 50% discounted prices. That could well be the future standard, common denominator, or starting point for US biosimilar pricing.
Biosimilar Nomenclature Issues Remain Unresolved
The FDA has yet to issue any guidance, much less regulation, concerning biosimilar product nomenclature. This particularly concerns the established, official/compendial nonproprietary product names used for designating prescriptions and marketing. Usually US Adopted Names (USANs, www.ama-assn.org/go/usan) these are meant to be a short, usually unique, and easily remembered. The unique names desired by innovator companies favor prescription-filling safety and tracking of adverse events, but they effectively require biosimilars to be branded and marketed, which raises their costs (also making interchangeability, which eliminates the need for marketing, more difficult). Using the same biosimilar/biogeneric product names, as with generic drugs, would favor lower costs and allow products to be sold with minimal or even no marketing, but confound safety and tracking. Thus it is an approach favored by biosimilar companies.
Other issues yet to be resolved include whether biosimilars should be identified as such in their names and whether the USAN/INN (the World Health Organization’s International Nonproprietary Name system [www.who.int/medicines/services/inn], which is the parent of USAN) nomenclature system should continue to be used, abandoned, or modified. Neither was ever designed to handle biopharmaceuticals. USAN/INN uses inherently generic active-agent–based names for both active agents and finished products. That works well enough for generic drugs, but not for biopharmaceuticals, for which it is simply too ambiguous and imprecise.
Both the biopharmaceutical industry and its customers need functional (descriptive) product and agent names rather than official names determined by laws and regulatory requirements, such as established names. There are as yet no unique, nonproprietary names suitable for these products, with federal regulators having resisted assigning unique names to biologics for the >100 years of their regulation.
The Biotechnology Information Institute recently filed a citizen petition with the US FDA requesting the following:
-
The FDA should assign both unique and biosimilar and/or biogeneric-type (nonunique) adequately descriptive names both for approved biologics and their active agents. The requested names would be based on rational science according to each product/entity (within the process = product paradigm) rather than constrained by regulatory requirements. They need not be official, just used in public documents related to review and approvals.
-
The agency should disclose sufficiently descriptive information needed for understanding what biopharmaceutical products and agents are, particularly concerning their bioprocessing and quality-related aspects. This should include sufficient information to enable meaningful tracking of product drift. FDA documents related to biologics approval and review now have essentially all their chemistry, manufacturing, and controls (CMC)–related information fully redacted (censored) such that the few relevant sentences included in product inserts/labeling are more informative than that documentation!
Further information about nomenclature issues and BII’s citizen petition can be found online at www.biopharmacopeia.com.
Will price reductions allow for sufficient profits? The margins will be less than for innovator products, but higher than for most generic drugs. Just a 5–10% market share of a $1 billion/year market is $50–100 million annually, which is more than the estimated total cost for developing most biosimilars. So even with fractured markets, these products will be profitable. Price competition and lower mark-ups (as for generic drugs) will best position those companies with portfolios of multiple biosimilars and/or larger portfolios of other products for marketing, bundled discounted sales, and overall profit.
Some companies view biosimilars as their entry into the US biopharmaceuticals market. Some may be more concerned with capturing market share and establishing a presence and name on the US market rather than profiting from products with inherently limited niche sales potential. For some small biotech and foreign companies, getting a biopharmaceutical approved for the US market will be an invaluable achievement in itself (likely adding hundreds of millions of dollars to valuations and/or raising stock prices), which could be more important than actual sales. Other factors could spoil the market for biosimilars. For example, biobetters being “better” in some respects could capture a dominant market share if priced comparably to biosimilars. New me-too and fully innovative products for the same indications will take market share as well.
Companies that are best able to compete will be those that invest in developing the most cost-effective bioprocessing. Some may license and use the latest technologies; others may use legacy, off-patent bioprocess methods, even emulating a decades-old reference product’s legacy manufacturing process, with associated higher costs of goods. Wherever feasible, most biosimilar developers are scaling-up and planning for commercial manufacture with single-use systems. So in many respects, biosimilar development is promoting bioprocess innovation.
No matter how the US biosimilar (and biobetter) market evolves, these products and companies will come to numerically dominate the biopharmaceutical industry. They could quickly double or triple the number of biopharmaceuticals on the market. And that will make it even more critical to resolve a number of regulatory issues such as product nomenclature.
How Many Products Are in the Pipeline?
The BII pipeline database currently includes 514 candidate biosimilars and 402 biobetters, a total of 916 products in development concerning 119 of the >140 recombinant proteins that are currently approved in the United States. Compare that number with the 907 products the Pharmaceutical Research and Manufacturers’ Association (PhRMA) reports for the US biopharmaceutical (originators) clinical development pipeline (8).
Table 2 lists some current biosimilars and biobetters in development according to their reference products (with recent annual sales) and product class. As with most pharmaceutical pipelines, the majority of products represented are in preclinical stages; as is normal, most will fail or be abandoned along the way. We can assume higher trial entry and approval rates for biosimilars than for innovative products, but the truth remains to be seen.
Candidate products have yet to be disclosed from many likely major biosimilar players, including some of the largest international pharmaceutical and generic drug companies, so they are not represented in the BII database. A number of those companies probably will be licensing-in products that are now being developed by other (smaller), companies. Otherwise, the BII analyses include every company known to be active in developing biosimilars and/or biobetters.
Where are biosimilars in the pipeline? Very few biosimilars in development have yet entered clinical trials to support US, EU, or other highly regulated country approvals. Almost half of the 514 biosimilar candidates (232) are in preclinical stages. With the exception of EU-approved products, most “marketed” biosimilars are essentially biogenerics in minimally regulated international commerce (needing new trials for major-market approval). Thus, you can consider ≥80% of marketed biosimilars to be at the preclinical stage in terms of US and EU markets. Relatively few biosimilars are in clinical trials overall, particularly compared with biobetters, which can be thought of as nothing new, simply business as usual (partially innovative, next-generation, me-too products).
So far, biosimilar development has been orderly in general, with no desperation or companies racing to be first into trials or to file for approvals. Few developers are rushing to get trials done early for major targets. A few large companies that took early leads in doing so (before any useful FDA guidance) have recently suspended their efforts. For example, Celltrion, Teva, and Samsung have halted trials of biosimilar rituximab (Rituxan from Genentech and Biogen Idec).
Unlike generic drugs, for which the first US approval receives six months of market exclusivity, biosimilars get no reward for being first to market. In fact, the first companies to file will probably bear the brunt of resolving patent disputes, which could cost tens of millions of dollars, allowing products filed/approved later to avoid much of that trouble. The first to file also will probably have to face more regulatory hurdles. An unknown number of companies may be holding off or proceeding slowly as they look forward to future interchangeable approvals for major market countries — or at least for better guidance from the FDA. With most of the best reference targets coming off patent in coming years, developers still have time to organize trials for the most target-worthy ones.
A large number of biosimilars will enter trials in the next few years. Such studies can be completed relatively quickly because they will usually be comparative bioequivalence trials only rather than full phase 3–type trials for proving efficacy and safety. With some reference products, however, biosimilar trials will be expensive and complex, including treatments of cancer, arthritis, and other complex, chronic diseases for which purchasing reference products for comparison alone can easily cost tens of millions of dollars. Just purchasing those reference products for testing can be a challenge. Some are simply not available (that is, sold only by their manufacturers, which have no obligation to sell products to potential competitors). It may be difficult for some developers to find trial sites and specialists that are not already under contract or don’t have conflicts of interest from working with reference-product companies. The ability to conduct trials probably will be the main choke-point in biosimilar development. Those companies that do not reserve contract research organizations (CROs) and trial sites ahead of the pack may encounter problems later on. Who’s Developing Biosimilars?
Figure 3 shows the geographic distribution of companies involved in development of biosimilars and biobetters, and Figure 4 shows the geographic distribution of the associated products in development. Asia and the Pacific Rim collectively have the most biosimilar companies and products in development, followed by the United States and Europe. It remains to be seen how many Asian and other foreign products will ultimately enter US-oriented clinical trials. Far ahead in innovative biopharmaceutical development, the United States is also the clear leader in terms of developing incrementally innovative biobetter products.
Individual companies in different regions have different numbers of products in development. On average, Asian companies claim roughly about twice as many biosimilars in development compared with their US counterparts, which in turn have about twice as many biobetters in development. What Asian companies report as a product in development, however, may be different in the level of effort involved than those based in the United States and elsewhere. Table 3 lists companies with the most biosimilar products (≥10) reported in development.
With by far the largest portfolio, Harvest Moon Pharmaceuticals USA Inc. (one of the very first biosimilar developers) is an outlier, and it is so far the only US company with ≥10 biosimilars reported in development. Sooner or later, however, many of the largest US and European companies will be disclosing their development portfolios, including the most likely future major players (particularly in terms of marketing) with ≥10 candidates.
Figure 5 indicates the size of companies that have reported biosimilars (160 total) or biobetters (173 total) in development as well as the total number of companies involved (including those not yet disclosing their pipeline products). The distinct lack of established, public, and midsized US biotechnology companies among biosimilar developers is not apparent in the figure. But those companies are generally preoccupied with innovative product development and biobetters.
Figure 5:
How Will the US Market Evolve?
With few biosimilars in trials so far, it is too early to attempt to predict the likely size and diversity of the US market. However, it is becoming clear that a large number of biosimilars (and biobetters) will enter the market as their reference product patents expire. With many new entrants to the industry, bioprocessing equipment and services sales have increased already. Contract manufacturing organizations (CMOs) report an overall 15% increase in business from biosimilars. Major early milestones (and barriers) for all biosimilar development programs will include reference-product testing (easily costing >$1 million) using such methods as mass spectrometry; bioprocess development (with CMOs commonly quoting ∼$3 million to get to hundreds of liters in full CGMP compliance); and clinical trials (minimally costing millions of dollars and the most expensive effort).
Biosimilars will lead to biopharmaceutical revenue contraction, at least for the competing products involved. Because of increased competition as products come off patent and follow-ons enter the market, the market size will contract for each reference product and its biosimilars, biobetters, and other me-too products as they all compete, presumably at lower prices. Therefore, although biosimilars are very much expanding the market in numbers of players and products, the overall size of the markets for each group of affected products will contract as less-expensive versions gain traction.
BII believes that the US biosimilar market will be more like that of generic rather than innovator drugs. Much as the blockbuster asthma/allergy drug Singulair (montelukast sodium) recently went generic with 10 products approved by the FDA on the same day, some 10 or more biosimilars could rapidly enter the market for each successful reference product. Applying that to the 40 reference products with current ≥$1 billion/year sales suggests ≥400 biosimilars entering the market.Presuming that just five biosimilars enter the market for the 18 products with sales ≥$500 million, that’s an additional 90 products — for a total of ∼500 follow-on biopharmaceuticals entering the US market in this decade alone!
And that is probably a low-ball estimate, with more products and players potentially involved. If for each major reference product, for example, biosimilars are launched by just three Big Pharma companies, three smaller biotech companies, three of the largest generic drug companies, and three foreign new entrants to the US market, then that equates to a dozen biosimilars for one reference product. Those numbers are likely to be higher in reality, with most of the largest Big Pharma and generic companies expected to add biosimilars to round out their portfolios sooner or later. In other words, there will be a large amount of competition.
What types of companies will make up the US biosimilars market? All types and sizes from everywhere will be represented, but most of those involved will tend to be either quite large or quite small. Will too many players make for too much competition? Maybe so — especially for those companies expecting high, innovator-like, profit margins for biosimilars.
What will pricing models look like? The current consensus is based on limited EU experience and projects biosimilars entering the US market to be discounted 20–30% relative to their reference products. But BII believes that is too low an estimate for a market where insurers/payers will rapidly switch to biosimilar use, forcing companies into price competition (as with generic drugs). Already a future major player, Samsung Bioepis (the joint venture of Samsung with Biogen Idec) has reported that it intends to launch a full portfolio of biosimilars — including MAbs — in the United States at 50% discounted prices. That could well be the future standard, common denominator, or starting point for US biosimilar pricing.
Biosimilar Nomenclature Issues Remain Unresolved
The FDA has yet to issue any guidance, much less regulation, concerning biosimilar product nomenclature. This particularly concerns the established, official/compendial nonproprietary product names used for designating prescriptions and marketing. Usually US Adopted Names (USANs, www.ama-assn.org/go/usan) these are meant to be a short, usually unique, and easily remembered. The unique names desired by innovator companies favor prescription-filling safety and tracking of adverse events, but they effectively require biosimilars to be branded and marketed, which raises their costs (also making interchangeability, which eliminates the need for marketing, more difficult). Using the same biosimilar/biogeneric product names, as with generic drugs, would favor lower costs and allow products to be sold with minimal or even no marketing, but confound safety and tracking. Thus it is an approach favored by biosimilar companies.
Other issues yet to be resolved include whether biosimilars should be identified as such in their names and whether the USAN/INN (the World Health Organization’s International Nonproprietary Name system [www.who.int/medicines/services/inn], which is the parent of USAN) nomenclature system should continue to be used, abandoned, or modified. Neither was ever designed to handle biopharmaceuticals. USAN/INN uses inherently generic active-agent–based names for both active agents and finished products. That works well enough for generic drugs, but not for biopharmaceuticals, for which it is simply too ambiguous and imprecise.
Both the biopharmaceutical industry and its customers need functional (descriptive) product and agent names rather than official names determined by laws and regulatory requirements, such as established names. There are as yet no unique, nonproprietary names suitable for these products, with federal regulators having resisted assigning unique names to biologics for the >100 years of their regulation.
The Biotechnology Information Institute recently filed a citizen petition with the US FDA requesting the following:
-
The FDA should assign both unique and biosimilar and/or biogeneric-type (nonunique) adequately descriptive names both for approved biologics and their active agents. The requested names would be based on rational science according to each product/entity (within the process = product paradigm) rather than constrained by regulatory requirements. They need not be official, just used in public documents related to review and approvals.
-
The agency should disclose sufficiently descriptive information needed for understanding what biopharmaceutical products and agents are, particularly concerning their bioprocessing and quality-related aspects. This should include sufficient information to enable meaningful tracking of product drift. FDA documents related to biologics approval and review now have essentially all their chemistry, manufacturing, and controls (CMC)–related information fully redacted (censored) such that the few relevant sentences included in product inserts/labeling are more informative than that documentation!
Further information about nomenclature issues and BII’s citizen petition can be found online at www.biopharmacopeia.com.
Will price reductions allow for sufficient profits? The margins will be less than for innovator products, but higher than for most generic drugs. Just a 5–10% market share of a $1 billion/year market is $50–100 million annually, which is more than the estimated total cost for developing most biosimilars. So even with fractured markets, these products will be profitable. Price competition and lower mark-ups (as for generic drugs) will best position those companies with portfolios of multiple biosimilars and/or larger portfolios of other products for marketing, bundled discounted sales, and overall profit.
Some companies view biosimilars as their entry into the US biopharmaceuticals market. Some may be more concerned with capturing market share and establishing a presence and name on the US market rather than profiting from products with inherently limited niche sales potential. For some small biotech and foreign companies, getting a biopharmaceutical approved for the US market will be an invaluable achievement in itself (likely adding hundreds of millions of dollars to valuations and/or raising stock prices), which could be more important than actual sales. Other factors could spoil the market for biosimilars. For example, biobetters being “better” in some respects could capture a dominant market share if priced comparably to biosimilars. New me-too and fully innovative products for the same indications will take market share as well.
Companies that are best able to compete will be those that invest in developing the most cost-effective bioprocessing. Some may license and use the latest technologies; others may use legacy, off-patent bioprocess methods, even emulating a decades-old reference product’s legacy manufacturing process, with associated higher costs of goods. Wherever feasible, most biosimilar developers are scaling-up and planning for commercial manufacture with single-use systems. So in many respects, biosimilar development is promoting bioprocess innovation.
No matter how the US biosimilar (and biobetter) market evolves, these products and companies will come to numerically dominate the biopharmaceutical industry. They could quickly double or triple the number of biopharmaceuticals on the market. And that will make it even more critical to resolve a number of regulatory issues such as product nomenclature.
How Many Products Are in the Pipeline?
Candidate products have yet to be disclosed from many likely major biosimilar players, including some of the largest international pharmaceutical and generic drug companies, so they are not represented in the BII database. A number of those companies probably will be licensing-in products that are now being developed by other (smaller), companies. Otherwise, the BII analyses include every company known to be active in developing biosimilars and/or biobetters.
Where are biosimilars in the pipeline? Very few biosimilars in development have yet entered clinical trials to support US, EU, or other highly regulated country approvals. Almost half of the 514 biosimilar candidates (232) are in preclinical stages. With the exception of EU-approved products, most “marketed” biosimilars are essentially biogenerics in minimally regulated international commerce (needing new trials for major-market approval). Thus, you can consider ≥80% of marketed biosimilars to be at the preclinical stage in terms of US and EU markets. Relatively few biosimilars are in clinical trials overall, particularly compared with biobetters, which can be thought of as nothing new, simply business as usual (partially innovative, next-generation, me-too products).
So far, biosimilar development has been orderly in general, with no desperation or companies racing to be first into trials or to file for approvals. Few developers are rushing to get trials done early for major targets. A few large companies that took early leads in doing so (before any useful FDA guidance) have recently suspended their efforts. For example, Celltrion, Teva, and Samsung have halted trials of biosimilar rituximab (Rituxan from Genentech and Biogen Idec).
Unlike generic drugs, for which the first US approval receives six months of market exclusivity, biosimilars get no reward for being first to market. In fact, the first companies to file will probably bear the brunt of resolving patent disputes, which could cost tens of millions of dollars, allowing products filed/approved later to avoid much of that trouble. The first to file also will probably have to face more regulatory hurdles. An unknown number of companies may be holding off or proceeding slowly as they look forward to future interchangeable approvals for major market countries — or at least for better guidance from the FDA. With most of the best reference targets coming off patent in coming years, developers still have time to organize trials for the most target-worthy ones.
A large number of biosimilars will enter trials in the next few years. Such studies can be completed relatively quickly because they will usually be comparative bioequivalence trials only rather than full phase 3–type trials for proving efficacy and safety. With some reference products, however, biosimilar trials will be expensive and complex, including treatments of cancer, arthritis, and other complex, chronic diseases for which purchasing reference products for comparison alone can easily cost tens of millions of dollars. Just purchasing those reference products for testing can be a challenge. Some are simply not available (that is, sold only by their manufacturers, which have no obligation to sell products to potential competitors). It may be difficult for some developers to find trial sites and specialists that are not already under contract or don’t have conflicts of interest from working with reference-product companies. The ability to conduct trials probably will be the main choke-point in biosimilar development. Those companies that do not reserve contract research organizations (CROs) and trial sites ahead of the pack may encounter problems later on. Who’s Developing Biosimilars?
Figure 3 shows the geographic distribution of companies involved in development of biosimilars and biobetters, and Figure 4 shows the geographic distribution of the associated products in development. Asia and the Pacific Rim collectively have the most biosimilar companies and products in development, followed by the United States and Europe. It remains to be seen how many Asian and other foreign products will ultimately enter US-oriented clinical trials. Far ahead in innovative biopharmaceutical development, the United States is also the clear leader in terms of developing incrementally innovative biobetter products.
Individual companies in different regions have different numbers of products in development. On average, Asian companies claim roughly about twice as many biosimilars in development compared with their US counterparts, which in turn have about twice as many biobetters in development. What Asian companies report as a product in development, however, may be different in the level of effort involved than those based in the United States and elsewhere. Table 3 lists companies with the most biosimilar products (≥10) reported in development.
With by far the largest portfolio, Harvest Moon Pharmaceuticals USA Inc. (one of the very first biosimilar developers) is an outlier, and it is so far the only US company with ≥10 biosimilars reported in development. Sooner or later, however, many of the largest US and European companies will be disclosing their development portfolios, including the most likely future major players (particularly in terms of marketing) with ≥10 candidates.
Figure 5 indicates the size of companies that have reported biosimilars (160 total) or biobetters (173 total) in development as well as the total number of companies involved (including those not yet disclosing their pipeline products). The distinct lack of established, public, and midsized US biotechnology companies among biosimilar developers is not apparent in the figure. But those companies are generally preoccupied with innovative product development and biobetters.
Figure 5:
How Will the US Market Evolve?
With few biosimilars in trials so far, it is too early to attempt to predict the likely size and diversity of the US market. However, it is becoming clear that a large number of biosimilars (and biobetters) will enter the market as their reference product patents expire. With many new entrants to the industry, bioprocessing equipment and services sales have increased already. Contract manufacturing organizations (CMOs) report an overall 15% increase in business from biosimilars. Major early milestones (and barriers) for all biosimilar development programs will include reference-product testing (easily costing >$1 million) using such methods as mass spectrometry; bioprocess development (with CMOs commonly quoting ∼$3 million to get to hundreds of liters in full CGMP compliance); and clinical trials (minimally costing millions of dollars and the most expensive effort).
Biosimilars will lead to biopharmaceutical revenue contraction, at least for the competing products involved. Because of increased competition as products come off patent and follow-ons enter the market, the market size will contract for each reference product and its biosimilars, biobetters, and other me-too products as they all compete, presumably at lower prices. Therefore, although biosimilars are very much expanding the market in numbers of players and products, the overall size of the markets for each group of affected products will contract as less-expensive versions gain traction.
BII believes that the US biosimilar market will be more like that of generic rather than innovator drugs. Much as the blockbuster asthma/allergy drug Singulair (montelukast sodium) recently went generic with 10 products approved by the FDA on the same day, some 10 or more biosimilars could rapidly enter the market for each successful reference product. Applying that to the 40 reference products with current ≥$1 billion/year sales suggests ≥400 biosimilars entering the market.Presuming that just five biosimilars enter the market for the 18 products with sales ≥$500 million, that’s an additional 90 products — for a total of ∼500 follow-on biopharmaceuticals entering the US market in this decade alone!
And that is probably a low-ball estimate, with more products and players potentially involved. If for each major reference product, for example, biosimilars are launched by just three Big Pharma companies, three smaller biotech companies, three of the largest generic drug companies, and three foreign new entrants to the US market, then that equates to a dozen biosimilars for one reference product. Those numbers are likely to be higher in reality, with most of the largest Big Pharma and generic companies expected to add biosimilars to round out their portfolios sooner or later. In other words, there will be a large amount of competition.
What types of companies will make up the US biosimilars market? All types and sizes from everywhere will be represented, but most of those involved will tend to be either quite large or quite small. Will too many players make for too much competition? Maybe so — especially for those companies expecting high, innovator-like, profit margins for biosimilars.
What will pricing models look like? The current consensus is based on limited EU experience and projects biosimilars entering the US market to be discounted 20–30% relative to their reference products. But BII believes that is too low an estimate for a market where insurers/payers will rapidly switch to biosimilar use, forcing companies into price competition (as with generic drugs). Already a future major player, Samsung Bioepis (the joint venture of Samsung with Biogen Idec) has reported that it intends to launch a full portfolio of biosimilars — including MAbs — in the United States at 50% discounted prices. That could well be the future standard, common denominator, or starting point for US biosimilar pricing.
Biosimilar Nomenclature Issues Remain Unresolved
The FDA has yet to issue any guidance, much less regulation, concerning biosimilar product nomenclature. This particularly concerns the established, official/compendial nonproprietary product names used for designating prescriptions and marketing. Usually US Adopted Names (USANs, www.ama-assn.org/go/usan) these are meant to be a short, usually unique, and easily remembered. The unique names desired by innovator companies favor prescription-filling safety and tracking of adverse events, but they effectively require biosimilars to be branded and marketed, which raises their costs (also making interchangeability, which eliminates the need for marketing, more difficult). Using the same biosimilar/biogeneric product names, as with generic drugs, would favor lower costs and allow products to be sold with minimal or even no marketing, but confound safety and tracking. Thus it is an approach favored by biosimilar companies.
Other issues yet to be resolved include whether biosimilars should be identified as such in their names and whether the USAN/INN (the World Health Organization’s International Nonproprietary Name system [www.who.int/medicines/services/inn], which is the parent of USAN) nomenclature system should continue to be used, abandoned, or modified. Neither was ever designed to handle biopharmaceuticals. USAN/INN uses inherently generic active-agent–based names for both active agents and finished products. That works well enough for generic drugs, but not for biopharmaceuticals, for which it is simply too ambiguous and imprecise.
Both the biopharmaceutical industry and its customers need functional (descriptive) product and agent names rather than official names determined by laws and regulatory requirements, such as established names. There are as yet no unique, nonproprietary names suitable for these products, with federal regulators having resisted assigning unique names to biologics for the >100 years of their regulation.
The Biotechnology Information Institute recently filed a citizen petition with the US FDA requesting the following:
-
The FDA should assign both unique and biosimilar and/or biogeneric-type (nonunique) adequately descriptive names both for approved biologics and their active agents. The requested names would be based on rational science according to each product/entity (within the process = product paradigm) rather than constrained by regulatory requirements. They need not be official, just used in public documents related to review and approvals.
-
The agency should disclose sufficiently descriptive information needed for understanding what biopharmaceutical products and agents are, particularly concerning their bioprocessing and quality-related aspects. This should include sufficient information to enable meaningful tracking of product drift. FDA documents related to biologics approval and review now have essentially all their chemistry, manufacturing, and controls (CMC)–related information fully redacted (censored) such that the few relevant sentences included in product inserts/labeling are more informative than that documentation!
Further information about nomenclature issues and BII’s citizen petition can be found online at www.biopharmacopeia.com.
Will price reductions allow for sufficient profits? The margins will be less than for innovator products, but higher than for most generic drugs. Just a 5–10% market share of a $1 billion/year market is $50–100 million annually, which is more than the estimated total cost for developing most biosimilars. So even with fractured markets, these products will be profitable. Price competition and lower mark-ups (as for generic drugs) will best position those companies with portfolios of multiple biosimilars and/or larger portfolios of other products for marketing, bundled discounted sales, and overall profit.
Some companies view biosimilars as their entry into the US biopharmaceuticals market. Some may be more concerned with capturing market share and establishing a presence and name on the US market rather than profiting from products with inherently limited niche sales potential. For some small biotech and foreign companies, getting a biopharmaceutical approved for the US market will be an invaluable achievement in itself (likely adding hundreds of millions of dollars to valuations and/or raising stock prices), which could be more important than actual sales. Other factors could spoil the market for biosimilars. For example, biobetters being “better” in some respects could capture a dominant market share if priced comparably to biosimilars. New me-too and fully innovative products for the same indications will take market share as well.
Companies that are best able to compete will be those that invest in developing the most cost-effective bioprocessing. Some may license and use the latest technologies; others may use legacy, off-patent bioprocess methods, even emulating a decades-old reference product’s legacy manufacturing process, with associated higher costs of goods. Wherever feasible, most biosimilar developers are scaling-up and planning for commercial manufacture with single-use systems. So in many respects, biosimilar development is promoting bioprocess innovation.
No matter how the US biosimilar (and biobetter) market evolves, these products and companies will come to numerically dominate the biopharmaceutical industry. They could quickly double or triple the number of biopharmaceuticals on the market. And that will make it even more critical to resolve a number of regulatory issues such as product nomenclature.
Individual companies in different regions have different numbers of products in development. On average, Asian companies claim roughly about twice as many biosimilars in development compared with their US counterparts, which in turn have about twice as many biobetters in development. What Asian companies report as a product in development, however, may be different in the level of effort involved than those based in the United States and elsewhere. Table 3 lists companies with the most biosimilar products (≥10) reported in development.
With by far the largest portfolio, Harvest Moon Pharmaceuticals USA Inc. (one of the very first biosimilar developers) is an outlier, and it is so far the only US company with ≥10 biosimilars reported in development. Sooner or later, however, many of the largest US and European companies will be disclosing their development portfolios, including the most likely future major players (particularly in terms of marketing) with ≥10 candidates.
Figure 5 indicates the size of companies that have reported biosimilars (160 total) or biobetters (173 total) in development as well as the total number of companies involved (including those not yet disclosing their pipeline products). The distinct lack of established, public, and midsized US biotechnology companies among biosimilar developers is not apparent in the figure. But those companies are generally preoccupied with innovative product development and biobetters.
Figure 5:
How Will the US Market Evolve?
With few biosimilars in trials so far, it is too early to attempt to predict the likely size and diversity of the US market. However, it is becoming clear that a large number of biosimilars (and biobetters) will enter the market as their reference product patents expire. With many new entrants to the industry, bioprocessing equipment and services sales have increased already. Contract manufacturing organizations (CMOs) report an overall 15% increase in business from biosimilars. Major early milestones (and barriers) for all biosimilar development programs will include reference-product testing (easily costing >$1 million) using such methods as mass spectrometry; bioprocess development (with CMOs commonly quoting ∼$3 million to get to hundreds of liters in full CGMP compliance); and clinical trials (minimally costing millions of dollars and the most expensive effort).
Biosimilars will lead to biopharmaceutical revenue contraction, at least for the competing products involved. Because of increased competition as products come off patent and follow-ons enter the market, the market size will contract for each reference product and its biosimilars, biobetters, and other me-too products as they all compete, presumably at lower prices. Therefore, although biosimilars are very much expanding the market in numbers of players and products, the overall size of the markets for each group of affected products will contract as less-expensive versions gain traction.
BII believes that the US biosimilar market will be more like that of generic rather than innovator drugs. Much as the blockbuster asthma/allergy drug Singulair (montelukast sodium) recently went generic with 10 products approved by the FDA on the same day, some 10 or more biosimilars could rapidly enter the market for each successful reference product. Applying that to the 40 reference products with current ≥$1 billion/year sales suggests ≥400 biosimilars entering the market.Presuming that just five biosimilars enter the market for the 18 products with sales ≥$500 million, that’s an additional 90 products — for a total of ∼500 follow-on biopharmaceuticals entering the US market in this decade alone!
And that is probably a low-ball estimate, with more products and players potentially involved. If for each major reference product, for example, biosimilars are launched by just three Big Pharma companies, three smaller biotech companies, three of the largest generic drug companies, and three foreign new entrants to the US market, then that equates to a dozen biosimilars for one reference product. Those numbers are likely to be higher in reality, with most of the largest Big Pharma and generic companies expected to add biosimilars to round out their portfolios sooner or later. In other words, there will be a large amount of competition.
What types of companies will make up the US biosimilars market? All types and sizes from everywhere will be represented, but most of those involved will tend to be either quite large or quite small. Will too many players make for too much competition? Maybe so — especially for those companies expecting high, innovator-like, profit margins for biosimilars.
What will pricing models look like? The current consensus is based on limited EU experience and projects biosimilars entering the US market to be discounted 20–30% relative to their reference products. But BII believes that is too low an estimate for a market where insurers/payers will rapidly switch to biosimilar use, forcing companies into price competition (as with generic drugs). Already a future major player, Samsung Bioepis (the joint venture of Samsung with Biogen Idec) has reported that it intends to launch a full portfolio of biosimilars — including MAbs — in the United States at 50% discounted prices. That could well be the future standard, common denominator, or starting point for US biosimilar pricing.
Biosimilar Nomenclature Issues Remain Unresolved
The FDA has yet to issue any guidance, much less regulation, concerning biosimilar product nomenclature. This particularly concerns the established, official/compendial nonproprietary product names used for designating prescriptions and marketing. Usually US Adopted Names (USANs, www.ama-assn.org/go/usan) these are meant to be a short, usually unique, and easily remembered. The unique names desired by innovator companies favor prescription-filling safety and tracking of adverse events, but they effectively require biosimilars to be branded and marketed, which raises their costs (also making interchangeability, which eliminates the need for marketing, more difficult). Using the same biosimilar/biogeneric product names, as with generic drugs, would favor lower costs and allow products to be sold with minimal or even no marketing, but confound safety and tracking. Thus it is an approach favored by biosimilar companies.
Other issues yet to be resolved include whether biosimilars should be identified as such in their names and whether the USAN/INN (the World Health Organization’s International Nonproprietary Name system [www.who.int/medicines/services/inn], which is the parent of USAN) nomenclature system should continue to be used, abandoned, or modified. Neither was ever designed to handle biopharmaceuticals. USAN/INN uses inherently generic active-agent–based names for both active agents and finished products. That works well enough for generic drugs, but not for biopharmaceuticals, for which it is simply too ambiguous and imprecise.
Both the biopharmaceutical industry and its customers need functional (descriptive) product and agent names rather than official names determined by laws and regulatory requirements, such as established names. There are as yet no unique, nonproprietary names suitable for these products, with federal regulators having resisted assigning unique names to biologics for the >100 years of their regulation.
The Biotechnology Information Institute recently filed a citizen petition with the US FDA requesting the following:
- The FDA should assign both unique and biosimilar and/or biogeneric-type (nonunique) adequately descriptive names both for approved biologics and their active agents. The requested names would be based on rational science according to each product/entity (within the process = product paradigm) rather than constrained by regulatory requirements. They need not be official, just used in public documents related to review and approvals.
- The agency should disclose sufficiently descriptive information needed for understanding what biopharmaceutical products and agents are, particularly concerning their bioprocessing and quality-related aspects. This should include sufficient information to enable meaningful tracking of product drift. FDA documents related to biologics approval and review now have essentially all their chemistry, manufacturing, and controls (CMC)–related information fully redacted (censored) such that the few relevant sentences included in product inserts/labeling are more informative than that documentation!
Further information about nomenclature issues and BII’s citizen petition can be found online at www.biopharmacopeia.com.
Will price reductions allow for sufficient profits? The margins will be less than for innovator products, but higher than for most generic drugs. Just a 5–10% market share of a $1 billion/year market is $50–100 million annually, which is more than the estimated total cost for developing most biosimilars. So even with fractured markets, these products will be profitable. Price competition and lower mark-ups (as for generic drugs) will best position those companies with portfolios of multiple biosimilars and/or larger portfolios of other products for marketing, bundled discounted sales, and overall profit.
Some companies view biosimilars as their entry into the US biopharmaceuticals market. Some may be more concerned with capturing market share and establishing a presence and name on the US market rather than profiting from products with inherently limited niche sales potential. For some small biotech and foreign companies, getting a biopharmaceutical approved for the US market will be an invaluable achievement in itself (likely adding hundreds of millions of dollars to valuations and/or raising stock prices), which could be more important than actual sales. Other factors could spoil the market for biosimilars. For example, biobetters being “better” in some respects could capture a dominant market share if priced comparably to biosimilars. New me-too and fully innovative products for the same indications will take market share as well.
Companies that are best able to compete will be those that invest in developing the most cost-effective bioprocessing. Some may license and use the latest technologies; others may use legacy, off-patent bioprocess methods, even emulating a decades-old reference product’s legacy manufacturing process, with associated higher costs of goods. Wherever feasible, most biosimilar developers are scaling-up and planning for commercial manufacture with single-use systems. So in many respects, biosimilar development is promoting bioprocess innovation.
No matter how the US biosimilar (and biobetter) market evolves, these products and companies will come to numerically dominate the biopharmaceutical industry. They could quickly double or triple the number of biopharmaceuticals on the market. And that will make it even more critical to resolve a number of regulatory issues such as product nomenclature.
Author Details
Ronald A. Rader is author/publisher of BIOPHARMA: Biopharmaceutical Products in the US and European Markets (www.biopharma.com) and president of the Biotechnology Information Institute, 1700 Rockville Pike, Suite 400, Rockville, MD 20852; 1-301-424-0255; biotech@biopharma.com.
REFERENCES
1.) Rader, RA. 2007. What Is a Generic Biopharmaceutical? Biogeneric? Follow-On Protein? Biosimilar? Follow-On Biologic? Part 1: Introduction and Basic Paradigms. BioProcess Int. 5:28-38.
2.) Rader, RA. 2007. What Is a Generic Biopharmaceutical? Biogeneric? Follow-On Protein? Biosimilar? Follow-On Biologic? Part 2: Information, Nomenclature, Perceptions, and the Market. BioProcess Int. 5:20-38. 3.) Rader, RA. 2008. (Re)Defining Biopharmaceutical?. Nat. Biotechnol. 26:743-751. 4.) Rader, RA. 2012.Charting the Biosimilar and Biobetter Development Pipeline, FirstWord Pharma, London. 5.) Rader, RA.. 6.) Rader, RA.. 7.).