WWW.JUPITERIMAGES.COM
Viral safety is required for biologics manufactured to treat human diseases. Although significant improvements in ensuring viral safety have been made over the past few decades, “zero risk” of viral contamination is a myth. Viral contamination risk can be carefully managed by screening raw materials, testing process intermediates, and evaluating how effectively manufacturing processes remove and inactivate viruses.
Viral clearance studies verify virus removal or inactivation by a manufacturing process. Although regulatory agencies have expectations for the designs of those studies, no standard expectations for clearance levels apply to every product. Just as each biologic and the raw materials and manufacturing process used to generate it are unique, so too are the risks of viral contamination and viral clearance expectations.
A product’s expected level of clearance depends on the potential viral contaminant load in its source materials. That level incorporates additional clearance for an assurance of safety in a final-product dose. To evaluate the viral clearance capacity of a manufacturing process, the virus-reduction capacity of each unit operation is independently determined. That is done by spiking viruses into the preprocessed intermediate and comparing the amount of virus in the preprocessed load with that in the intermediate after processing. Individual spiking experiments are performed for each virus using an appropriately selected panel of viruses.
Part one of this series reviewed risks associated with viral contamination (1). It covered fundamental strategies, regulatory considerations in the design of viral clearance studies, and technologies for establishing a clear and transparent process for identifying and evaluating viral contamination risks. The conclusion here reviews some of the most commonly used technologies for inactivating and removing viruses.
The Need for Clearance
Despite rigorous controls with all biopharmaceutical products — including monoclonal antibodies (MAbs), recombinant proteins (RPs), human-plasma–derived products, and products derived from human and animal tissues (e.g., collagen) — source materials and adventitious viruses introduced during production present viral contamination risks. Source materials can include human plasma, cell lines, and human and animal tissue. The risk of viral contamination is higher for human- and animal-derived source materials than for nonbiological materials. Most important, viral contamination can have potential consequences with serious clinical and economic implications. Thus, viral clearance studies are a vital component of biopharmaceutical manufacturing processes evaluations and are designed to ensure that if a viral contaminant is present, then it will be cleared.
Sources of Viral Contamination
Historically, viral contamination of biologics has lead to the transmission of infectious virus to patients, many times with tragic consequences. For example, before implementation of viral inactivation procedures (e.g, solvent/detergent treatment), hepatitis C virus and human immunodeficiency virus type 1 were transmitted to patients through some human-plasma–derived biological products (1, 2). With sensitive plasma-screening procedures and viral clearance steps for related manufacturing processes, those products are now relatively safe (3, 4).
Biotechnology has increased the safety of biologics even further. To date, no infectious virus has been transmitted to a patient by a biopharmaceutical derived from a cell line. Occasionally, however, viral contamination has been detected in process intermediates (e.g., bulk harvests). Such contaminants were detected long before products reached patients, so no virus transmission to a patient occurred. Even so, the events caused great expenses for manufacturers and even product shortages. Detected viral contaminants in Chinese hamster ovary (CHO) cell processes have included murine minute virus (MMV), reovirus, Cache Valley virus (CVV), and vesivirus 2117 (5).
Viral contamination can occur through source materials or by the introduction of adventitious viruses during manufacturing. Source materials (e.g., cell substrates) can become contaminated a number of ways, including from sources derived from infected animals and use of contaminated cell culture components (e.g., bovine serum or porcine-derived trypsin). Adventitious viruses can be introduced into a manufacturing process through, for example, handling of cell cultures or media and use of contaminated biologics or nonbiologic reagents.
Scaling Down for Viral Clearance Studies
Biotechnology manufacturing is based on a series of purification steps. Manufacturers are responsible for identifying unit operations that have the potential to inactivate or remove viruses. They must then generate data to support the clearance potential of those unit operations. Assessment of the viral clearance potential of a manufacturing process as a whole can be achieved by considering the viral clearance performance of its individual unit operations. Therefore, each step to be evaluated is “spiked,” or challenged with the appropriate preparation of a high-titer test virus.
To evaluate the viral clearance potential of a manufacturing process, unit operations are selected that may provide virus inactivation or removal. Viral clearance studies are performed in virology laboratories that are separate from manufacturing facilities to prevent contamination of manufacturing processes. For safety reasons, these studies are performed using scaled-down models representing actual manufacturing processes and production systems. It is necessary, however, to demonstrate the validity of those scaled-down processes. Even for early stage studies, manufacturers must generate data to verify that downscaled processes accurately reflect full-scale manufacturing processes. Buffers, linear flow rates, contact times, and all other process parameters must represent those in full-scale processes. And product and impurity profiles must reflect full-scale processes. Unless scaled-down processes accurately mimic full-scale manufacturing processes, generated viral clearance data cannot be considered to be valid.
For scaled-down studies, a panel of viruses is selected to reflect potential viral contaminants in source materials. For a given unit operation, in general, each virus of the selected panel is independently spiked into preprocessed intermediate material (although multispike studies have been conducted). Analysts then perform the process step and determine the quantities of virus that are present in the process intermediates before and after processing.
Viral reduction levels are calculated by comparing the amount of virus in a preprocessed load material to that in a postprocessed sample. The level is typically expressed in terms of the logarithm (log10) of the reduction. Log viral reduction levels for each process unit operation are then summed, and analysts determine a cumulative process reduction value for each virus in the evaluated virus panel. Their results must confirm that the virus has been removed or inactivated and that the process includes excess capacity for viral clearance (6).
Regulatory Considerations
The range of viral clearance data required for biologics in the early stages of development is reduced in scope compared with that required for supporting product licensure. Viral clearance data must be generated to support the safety of products for clinical trials. Biologics manufactured for early stage clinical trials are typically manufactured using a process that has just been developed and where process parameter ranges are not well established. Consequently, robustness studies at the limits or outside of process parameter ranges are not required. When a worst-case parameter limit is known, however, it should be used for a viral clearance study; otherwise, the operating parameter target should be used. Similarly, chromatography resins used to manufacture early-stage clinical trial materials are generally quite new; therefore, no column sanitization studies are required, nor are studies with aged resins.
For products manufactured using well-characterized cell lines, the European Medicines Agency (EMA) requires a minimum evaluation of two orthogonal steps with a retrovirus and a parvovirus (7). Manufacturers must perform a risk assessment according to the ICH Q5A appendix. Based on the outcome of that assessment, evaluation of additional steps may be needed (1). For submissions to regulatory authorities other than the US Food and Drug Administration (FDA), manufacturers must conduct duplicate experiments to estimate reproducibility of viral clearance capabilities of their processes.
As product development proceeds toward product licensure, processing parameter ranges are established. For parameters that might affect viral clearance, robustness data should be generated to verify that viral clearance performance is not adversely affected throughout processing ranges. In addition, manufacturers should evaluate the efficacy of chromatography sanitization procedures to demonstrate that if an adventitious virus were introduced into a chromatography step, the column sanitization and regeneration procedures would prevent its carryover to subsequent runs. In addition, manufacturers must perform clearance studies with aged resins to confirm that viral clearance performance of each chromatography step does not deteriorate with extended resin use. Finally, if an abbreviated virus panel is used for early stage studies, that panel must be extended to show that viruses with varying characteristics can be cleared.
Clearance Methods
When developing a manufacturing process for a biopharmaceutical, the goal is to include unit operations that lead to good yields of highly purified protein product. Including steps that inactivate or remove viruses — made of proteins themselves — often involves an intricate balancing act: Potential viral contaminants must be cleared without jeopardizing the yield or purity of a product. An ideal situation is achieved when developmental viral clearance data can be generated to guide process development, but timelines and financial constraints often preclude this approach.
When possible, a manufacturing process should include unit operations that are specifically dedicated to virus inactivation or removal. For example, holding a process intermediate at a low pH (e.g., pH 3.6 or below) for a period of time or exposing process intermediates to detergent or a solvent–detergent mixture are unit operations that provide robust inactivation of enveloped viruses. Such steps can be shown to be robust; that is, they are effective over a range of varying conditions, such as the use of different buffers, pH levels, temperatures, concentrations, and exposure times.
Nonenveloped viruses are much more difficult to inactivate, and, often, conditions that will inactivate them are incompatible with a protein product. Reduction of such viruses typically depends on a removal step. Many manufacturing processes include virus- reduction filters because they are often transparent to a process, yet remove all viruses above a given size.
Most modern manufacturing processes include one or more chromatography unit operations. Although those steps are optimized to purify a protein, often they also provide some level of viral reduction. Many chromatography steps provide two to three logs of virus removal, but some manufacturers have optimized steps (e.g., anion-exchange chromatography) to provide very good viral clearance. However, chromatography is not a robust viral clearance method, because variations in pH, ionic strength, and other parameters can affect its viral clearance ability.
A Deeper Look at the Methods
To successfully mitigate risk, process engineers need a good understanding of the methods designed to either inactivate or remove viral contaminants (Table 1).
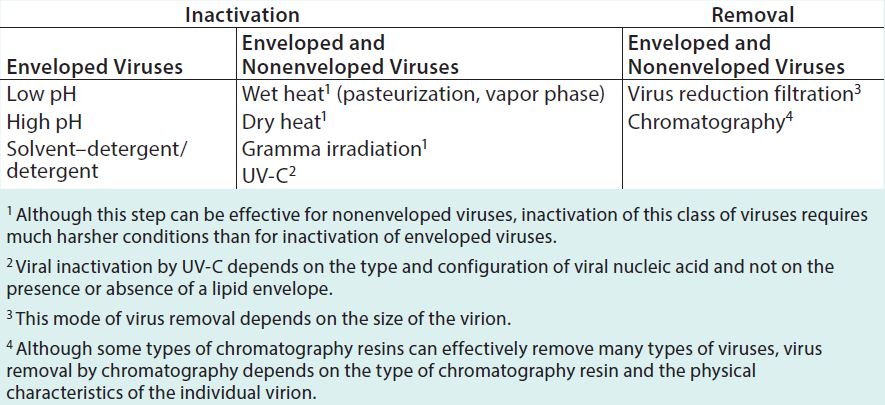
Table 1: Common methodologies that contribute to virus inactivation or removal in biopharmaceutical manufacturing processes
pH Treatment: Direct exposure of process intermediate to pH extremes has been used for viral clearance in biopharmaceutical manufacturing. For example, studies have proven that low pH treatment (e.g., pH 3.0–3.6) of MAbs following affinity chromatography is effective against enveloped viruses (8). High pH treatment using a sodium hydroxide solution is used to sanitize many chromatography columns, which can be effective against both enveloped and nonenveloped viruses (9–11). In general, exposure to pH extremes during manufacture of MAbs, plasma-derived products, products derived from tissue, recombinant proteins (RPs), and vaccines can provide effective, robust viral reduction (e.g., >4.0 log10).
Solvent/Detergent: Originally developed in the 1980s for use in manufacturing blood products, solvent/detergent (S/D) treatment is a proven method to inactivate enveloped viruses. Treatment of plasma-derived products with 0.3% tri(n-butyl) phosphate (TNBP)/0.2% sodium cholate (CA) can provide effective inactivation of infectious human immunodeficiency virus and hepatitis C virus (12). Today, solvent/ detergent or detergent alone is used in the manufacture of human-plasma– derived proteins and recombinant proteins. A detergent solubilizes a viral envelope’s lipid membrane structure. That disruption prevents a virus from binding to or infecting cells, thus rendering it inactive (13–14).
It is important to note that this process does not inactivate nonenveloped viruses. But because S/D targets lipids and lipid derivatives, this technique generally does not affect the potency of recombinant protein products (14–15).
Heat treatment, both wet (e.g., pasteurization, vapor heat) and dry, has been used for many human-plasma– derived and other animal-derived products (16–20). This method typically causes three-dimensional changes in the structure of viral proteins, rendering viruses nonfunctional or inactive. Although heat treatment can be effective against enveloped and nonenveloped viruses, some resistant, nonenveloped viruses (e.g., parvoviruses) require high temperatures to achieve effective inactivation, which may not be compatible with some protein products. High-heat treatment can change the structure of a protein product, leading to inactivation, reduced efficacy, or even immunogenicity and toxicity. Note that with dry heat treatment, the moisture content of a lyophilized cake might affect the efficacy of viral inactivation, making moisture control a critical parameter (21).
High-temperature short-time (HTST) treatment is an established method used in the food industry. It involves rapidly heating select materials to a predetermined temperature for a specified (short) time. This technique is gradually becoming an integral part of viral inactivation methodologies in the biopharmaceutical industry, particularly as an upstream risk-mitigation treatment for cell culture media (22–23).
HTST is thought to inactivate viral contaminants by denaturing proteins in the viral capsid (19). When performed at a sufficiently high temperature, this technique provides effective inactivation of resistant nonenveloped viruses (e.g., parvoviruses). A number of studies have further delineated the susceptibility of the parvovirus MMV (which is known to have contaminated mammalian cell manufacturing facilities) to heat by exposing virus-spiked cell culture media to a broad range of temperatures for various exposure times. The results of those studies indicate that HTST can inactivate MMV by three or more orders of magnitude. Thus, HTST is now recognized as a useful barrier technology for preventing adventitious contamination of mammalian cell culture processes in the biopharmaceutical industry.
Chromatography separates mixtures based on differences among the affinities of their components for a chromatographic resin matrix. Generally, this process is used to separate closely related molecules. As a unit operation, it is typically designed for purifying a protein product. Yet often, enveloped and nonenveloped viruses also can be separated from a product. Viral clearance depends on the physicochemical and biochemical properties of an individual virus. Removal of one type of virus does not necessarily mean that all viruses will be removed. In addition, chromatography buffers can inactivate enveloped viruses, such as low-pH buffers and buffers containing components that act as detergents. For cases in which enveloped viruses are inactivated, quantitative polymerase chain reaction (qPCR) can be used to assess virus removal.
Manufacturing processes for most biological products include chromatography operations. Most evaluations of viral clearance include assessment of at least one chromatography step. Robustness studies can verify that a chromatography operation provides consistent clearance over specified parameter operating ranges (24–27).
Many manufacturers use membrane chromatography steps in their processes. Chromatography membranes have the advantage of being disposable, which eliminates the need for sanitization or aged resin studies (28–29).
Virus Reduction Filtration: Many purification processes for biopharmaceuticals use virus-reduction filtration as an integral part of an overall strategy for viral clearance in upstream an downstream applications (30–31). Virus-reduction filters can provide robust and effective removal of large- and medium-sized viruses. Such filters also can effectively remove very small viruses (e.g., parvoviruses) with pore sizes ≤20 nm. But removal of such viruses can depend on manufacturing process parameters.
Gamma Irradiation: Irradiation using γ-rays is an accepted treatment for animal-derived materials. It has been effective in risk-mitigation strategies for raw materials and postpurification process intermediates. Viruses such as reovirus and CVV have been inactivated completely in frozen bovine serum using γ irradiation at 25–40 kGy (32). Irradiation of bovine serum is useful for mitigating the risk of viral contamination due to the presence of contaminants in unprocessed bulk biologics (e.g., CVV, reovirus, and epizootic hemorrhagic disease virus). However, this process must be strictly controlled to ensure consistent, reproducible, effective, and efficient virus inactivation.
Ultraviolet-C (UV-C) uses low-dose radiation at 254 nm to destroy viral nucleic acids while maintaining structural and functional integrity of manufactured proteins. Resistant parvoviruses have been shown to be much more susceptible to inactivation by UV-C than even enveloped viruses (33, 34). Researchers are exploring the application of this technology for treating cell culture media used for recombinant protein production as a means for mitigating viral contamination risk.
Making Viral Safety a Goal
Achieving zero risk of viral contamination in biologic production is impossible. But a strategy to ensure safety of biopharmaceuticals through risk mitigation involves careful choice and extensive testing of source materials; confirmed absence of contaminating infectious viruses in intermediates at relevant steps in a production process; and assessment of how well a manufacturing process inactivates and removes viruses. A manufacturing process designed with viral safety as a goal will include specific robust virus inactivation and removal steps as well as purification unit operations that provide further viral reduction. Although most manufacturers focus on incorporating viral-reduction steps into their downstream processes, including key viral clearance steps into an upstream process can further reduce the risk of viral contamination.
References
1 Gringeri A. Factor VIII Safety: Plasma-Derived versus Recombinant Products. Blood Transfus. 9, 2011: 366–370.
2 Bresee JS, et al. Hepatitis C Virus Infection Associated with Administration of Intravenous Immune Globulin: A Cohort Study. J. Am. Med. Assoc. 276(19) 1996: 1563–1567.
3 Horowitz B, et al. Viral Safety of Solvent-Detergent Treated Blood Products. Dev. Biol. Stand. 81, 1993: 147–161.
4 Bournouf T, Radosevich M. Reducing the Risk of Infection from Plasma Products: Specific Preventative Strategies. Blood Reviews 14(2) 2000: 94–110.
5 Wisher M. Virus Risk Mitigation for Raw Materials: A European Perspective. BioProcess Int. 11(9) 2013: 12–15.
6 Note for Guidance on Quality of Biotechnology Products: Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin (R1). US Fed. Reg. 63(185) 1998: 51074–51084.
7 EMEA/CHMP/BWP/398498/2005. Guideline on Virus Safety Evaluation of Biotechnological Investigational Medicinal Products. European Medicines Evaluation Agency: London, United Kingdom, 2008.
8 Brorson K, et al. Bracketed Generic Inactivation of Rodent Retroviruses By Low pH Treatment for Monoclonal Antibodies and Recombinant Proteins. Biotechnol. Bioeng. 82(3) 2003: 321–329.
9 Roberts PL. Virus Elimination During the Purification of Monoclonal Antibodies By Column Chromatography and Additional Steps. Biotechnol. Prog. 30(6) 2014: 1341–1347.
10 Pérez M, et al. Validation of Model Virus Removal and Inactivation Capacity of an Erythropoietin Purification Process. Biologicals 39(6) 2011: 430–437.
11 Boschetti N, et al. Stability of Minute Virus of Mice Against Temperature and Sodium Hydroxide. Biologicals 31(3) 2003: 181–185.
12 Prince AM, Horowitz B, Brotman B. Sterilisation of Hepatitis and HTLV-III Viruses by Exposure to Tri(n-butyl)phosphate and Sodium Cholate. Lancet 1(8483) 1986: 706–710.
13 Dichtelmüller HO, et al. Robustness of Solvent/Detergent Treatment of Plasma Derivatives: A Data Collection from Plasma Protein Therapeutics Association Member Companies. Transfusion 49(9) 2009: 1931–1943.
14 Hellstern P, Solheim BG. The Use of Solvent/Detergent Treatment in Pathogen Reduction of Plasma. Transfus. Med. Hemother. 38(1) 2011: 65–70.
15 Biesert L, Suhartono H. Solvent/ Detergent Treatment of Human Plasma: A Very Robust Method for Virus Inactivation. Validated Virus Safety of OCTAPLAS. Vox Sang. 74 Suppl. 1, 1998: 207–212.
16 Aghaie A, et al. Inactivation of Virus in Intravenous Immunoglobulin G Using Solvent/ Detergent Treatment and Pasteurization. Hum. Antibodies 17(3–4) 2008: 79–84.
17 Chandra S, Groener A, Feldman F. Effectiveness of Alternative Treatments for Reducing Potential Viral Contaminants from Plasma-Derived Products. Thromb. Res. 105(5) 2002: 391–400.
18 Barrett PN, et al. Inactivation of Hepatitis A Virus in Plasma Products By Vapor Heating. Transfusion 37(2) 1997: 215–220.
19 Mannucci PM, et al. The International Investigator Group: Low Risk of Viral Infection After Administration of Vapor- Heated Factor VIII Concentrate. Transfusion 32(2) 1992: 134–138.
20 Roberts PL, et al. Effect of Manufacturing Process Parameters on Virus Inactivation by Dry Heat Treatment at 80° C in Factor VIII. Vox Sang. 92(1) 2007: 56–63.
21 Savage M, et al. Determination of Adequate Moisture Content for Efficient Dry-Heat Viral Inactivation in Lyophilized Factor VIII By Loss on Drying and By Near Infrared Spectroscopy. Biologicals 26(2) 1998: 119–124.
22 Murphy M, Quesada GM, Chen D. Effectiveness of Mouse Minute Virus Inactivation By High-Temperature Short Time Treatment Technology: A Statistical Assessment. Biologicals 39(6) 2011: 438–443.
23 Schleh M et al. Susceptibility of Mouse Minute Virus to Inactivation By Heat in Two Cell Culture Media Types. Biotechnol. Prog. 25(3) 2009: 854–860.
24 Curtis S, et al. Generic/Matrix Evaluation of SV40 Clearance by Anion-Exchange Chromatography in Flow-Through Mode. Biotechnol. Bioeng. 84(2) 2003: 179–186.
25 Zhang M, et al. Quality by Design Approach for Viral Clearance By Protein A Chromatography. Biotechnol. Bioeng. 111(1) 2014: 95–103.
26 Strauss DM, et al. Anion-Exchange Chromatography Provides a Robust, Predictable Process to Ensure Viral Safety of Biotechnology Products. Biotechnol. Bioeng. 102(1) 2009: 168–175.
27 Strauss DM, et al. Strategies for Developing Design Spaces for Viral Clearance By Anion-Exchange Chromatography During Monoclonal Antibody Production. Biotechnol. Prog. 26(3) 2010: 750–755.
28 Zhou JX, et al. Viral Clearance Using Disposable Systems in Monoclonal Antibody Commercial Downstream Processing. Biotechnol. Bioeng. 100(3) 2008: 488–496.
29 Miesegaes GR, et al. Viral Clearance By Flow-Through Mode Ion-Exchange Columns and Membrane Adsorbers. Biotechnol. Prog. 30(1) 2014: 124–131.
30 Caballero S, et al. Robustness of Nanofiltration for Increasing the Viral Safety Margin of Biological Products. Biologicals 42(2) 2014: 79–85.
31 Marques BF, Roush DJ, Göklen KE. Virus Filtration of High-Concentration Monoclonal Antibody Solutions. Biotechnol. Prog. 25(2) 2005: 483–491.
32 Gauvin G, Nims R. Gamma-Irradiation of Serum for the Inactivation of Adventitious Contaminants. PDA J. Pharm. Sci. Technol. 64(5) 2010: 432–435.
33 Wang J, et al. Virus Inactivation and Protein Recovery in a Novel Ultraviolet-C Reactor. Vox Sang. 86(4) 2004: 230–238.
34 Yen S, et al. Treating Cell Culture Media with UV Irradiation Against Adventitious Agents: Minimal Impact on CHO Performance. Biotechnol. Prog. 30(5) 2014: 1190–1195.
Kathryn Martin Remington, PhD, is a principal scientist in clearance development services at BioReliance; kathy.remington@ bioreliance.com.
please guide the virus clearance from the monoclonal antibodies and spiking studies