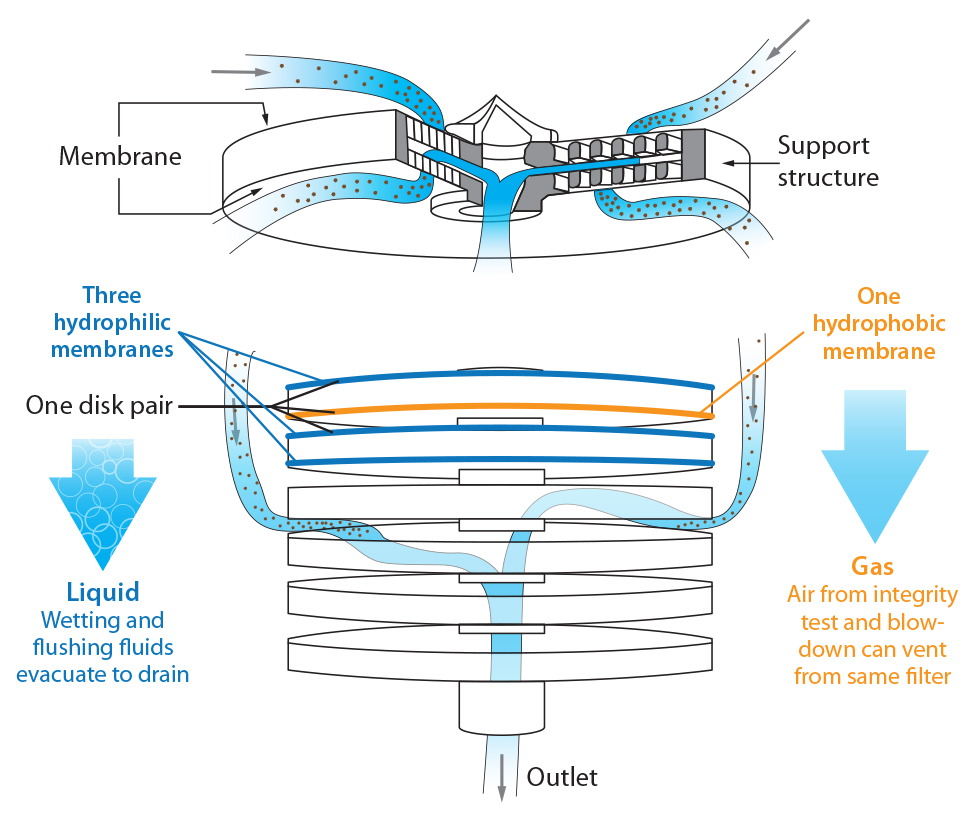
Figure 1: Design and layout of Millidisk and Millipak barrier filters
A number of regulatory guidelines recommend preuse integrity testing of critical sterilizing liquid filters for aseptic processing (1–3). Before sterilization, a preuse test will confirm that a filter is installed properly and was not damaged during shipment or handling. Performing a preuse test after sterilization detects damage that may have occurred during the sterilization cycle. Testing after sterilization limits risk, so it is a practice applied based on risk assessment. Because it is perceived to reduce business loss risk, preuse post-sterilization integrity testing (PUPSIT) is a current industry practice especially in manufacturing products that will be marketed in the European Union (EU).
Unfortunately, it can be difficult to perform a PUPSIT without breaching system sterility. A number of methods have been developed for running PUPSIT and performing line conditioning without compromising sterility. Such methods use a flush bag, catch-can/flush bottle, or filter arrangement to create a sterile boundary on the downstream side of the product filter. Some applications use the downstream hold tank as a sterile boundary.
Here we describe a robust and versatile approach to PUPSIT using a self-venting, all-in-one sterile barrier membrane filter from EMD Millipore, the life science business of Merck KGaA, Darmstadt, Germany, which operates as MilliporeSigma in the United States and Canada. We also include filtration line design considerations for implementing barrier filters.
Barrier Filters
Millipak and Millidisk barrier filters are stacked-disc devices that combine hydrophilic and hydrophobic sterilizing-grade Durapore membranes, both on the top and bottom of each disc (Figure 1). Because of that unique combination of different membranes in parallel configuration, the devices can filter condensate, steam, wetting liquid, and gases without compromising the sterility of a steam-sterilized, autoclaved, or gamma-irradiated system.
Barrier filters can be used downstream of sterilizing-grade filters to maintain system sterility. Water can pass through the hydrophilic discs during a flushing sequence; air can pass through the hydrophobic discs during integrity-test and drying sequences. A barrier filter acts as an automatic vent during testing and drying phases. The volume of particle-free water and air that can pass through these filters is unlimited.
Flushing and Testing Critical Product Filters
Although filter rewetting and retesting should remain an optional activity when preparing a product final filter (sterilizing-grade filter) in line, the PDA Technical Report 26 suggests up to three repetitions (3). The number of retests should be considered when sizing for a flush bag. Barrier filters could provide a more versatile solution.
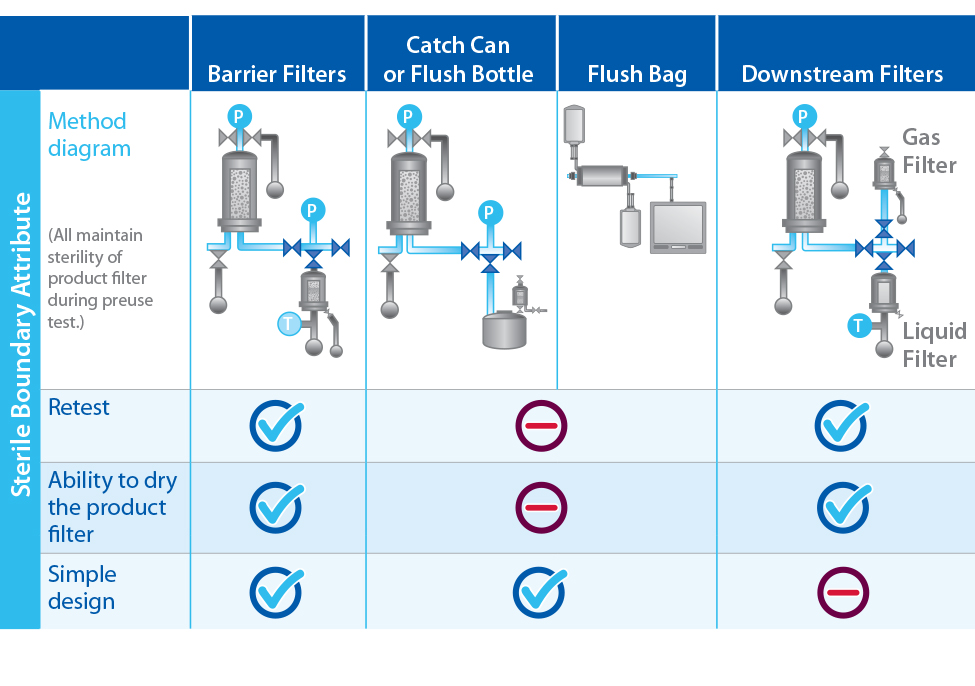
Figure 2: Examples of sterile boundary designs
Filters that are not wetted efficiently the first time could give false failed test results. If rewetting volume is limited, end users might discard integral filters that only marginally failed because of improper wetting. Doing so could lead to unnecessary quality investigations as well as downtime associated with setting up a system again before use. Using a barrier filter allows for rewetting and retesting with ease. Unlimited volumes of particle-free water can be filtered through them to wet these filters efficiently. Using a flush bag and/or catch-can instead creates a large footprint and could limit rewetting. Figure 2 summarizes the advantages and disadvantages of each sterile boundary method available.
Barrier filters also allow extractables flushing through a sterile boundary to drain. A barrier filter also can be used as a vent in system cooling after steam-in-place (SIP) sterilization and in filter drying after flushing to minimize product dilution.
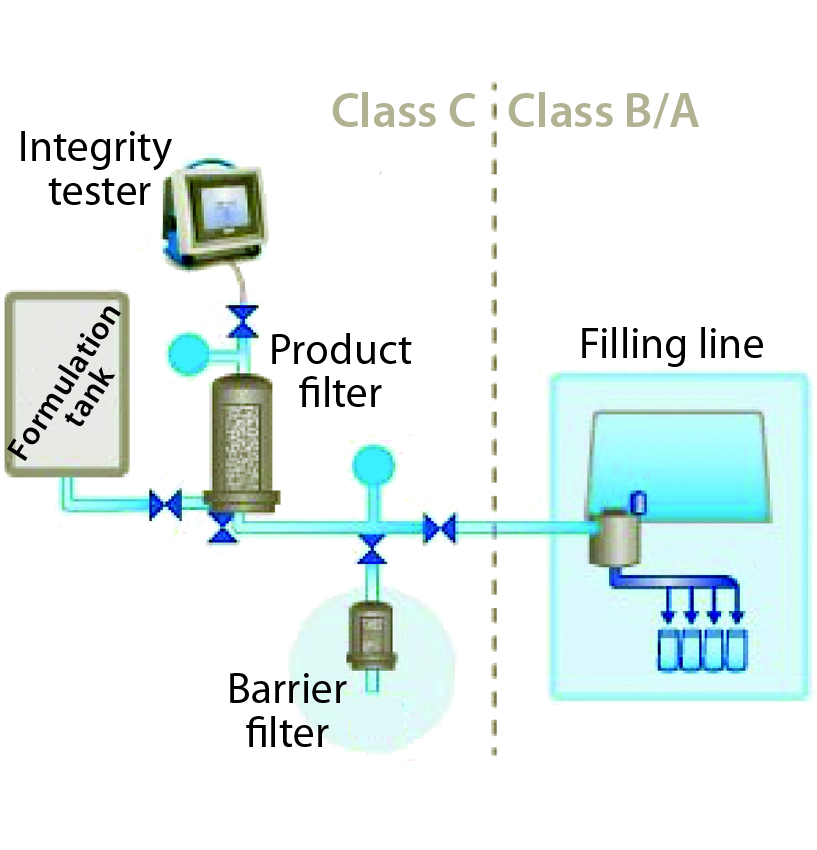
Figure 3: Barrier filters in a typical single filtration system (filling-line example)
Filtration Line Configuration and Operation
For critical filter applications such as final product-filling lines, barrier filters can be used to provide a sterile boundary while meeting regulatory requirements for preuse integrity testing. Typically, such filters are installed downstream of a product filter (Figure 3).
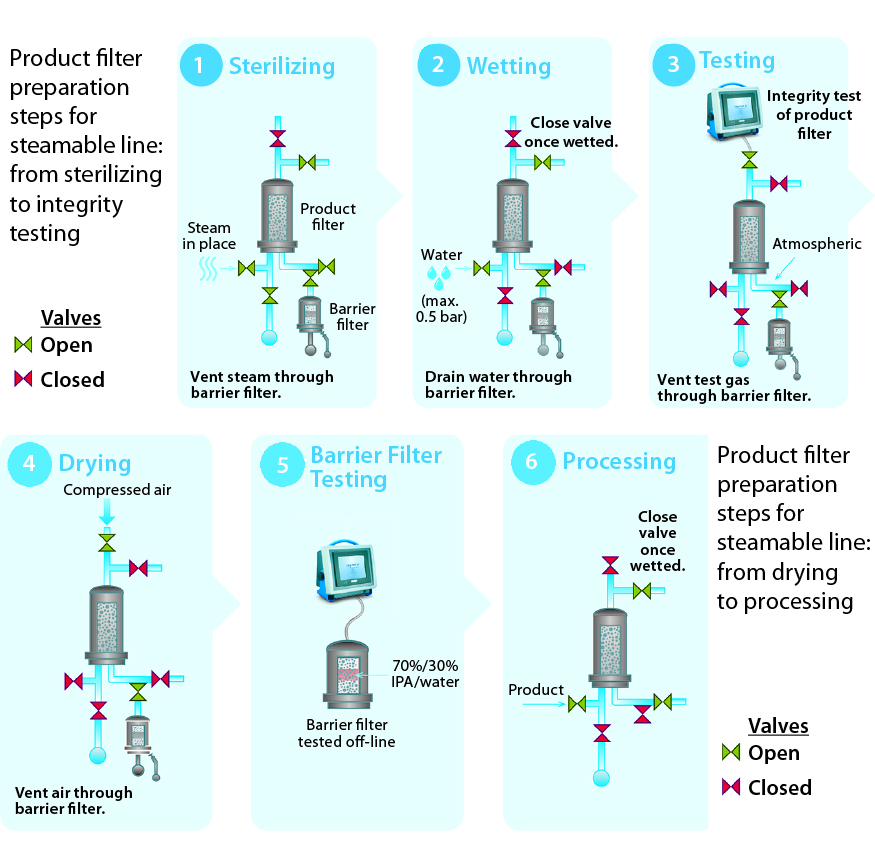
Figure 4: Product filter preparation steps for steamable line, from sterilizing to integrity testing
Figures 4 and 5 illustrate stepwise use of barrier filters to provide a sterile boundary in a stainless steel and a single-use filtration system with a single product filter. Regulators recommend redundant filtration as a risk-mitigation strategy for critical filtration applications. Redundant filtration is a type of serial filtration in which a second product filter is used as a back-up to protect against the possibility of an integrity failure for the primary product filter (3). Each filter must be independently integrity-testable in compliance with the relevant regulations or guidelines. The step-by-step approaches in Figures 4 and 5 can be applied to the second product filter in a redundant filtration system.
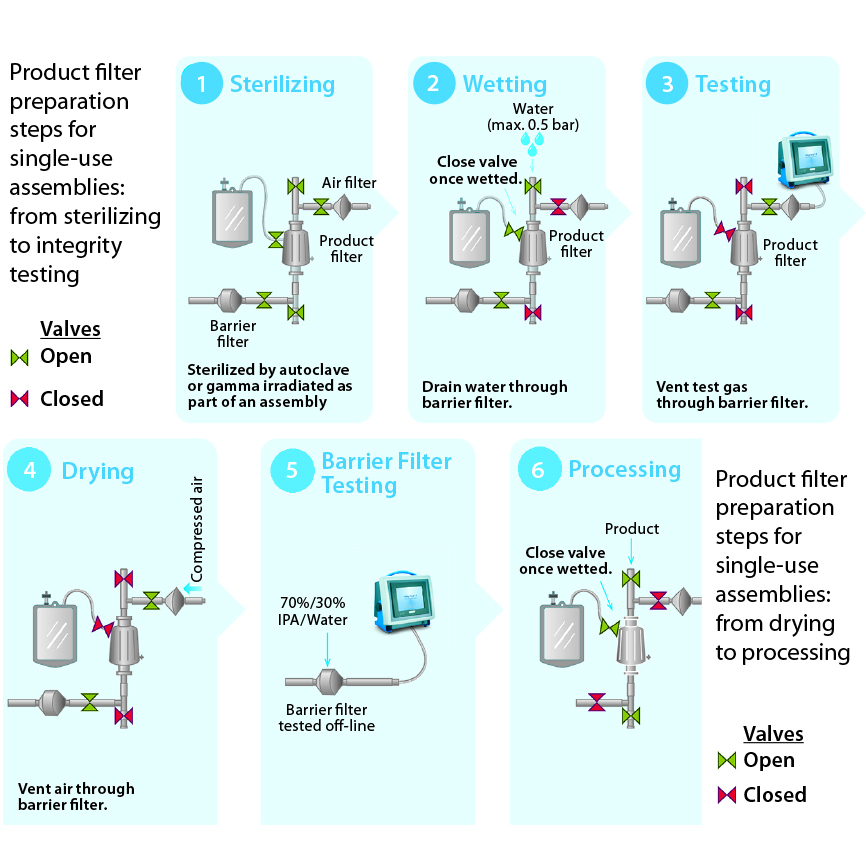
Figure 5: Product filter preparation steps for single-use assemblies, from sterilizing to integrity testing and from drying to process
Sterilizing: The process begins with sterilization of a filtration system by SIP, autoclaving, or gamma-irradiation. If the chosen method of sterilization is SIP, then the barrier filter’s low-point vent on its upstream side will be kept open during the SIP cycle. Condensate, steam, and air will pass through the filter to drain on its outlet. When the cycle ends, the low-point vent will be closed. The system then cools down with application of compressed gas (to maintain positive pressure as well as sterility in the filtrations system).
Wetting: The second step ensures that a product filter is totally wetted with particle-free water for its integrity test. This step also flushes away extractable residues from a sterilized product filter element. To keep a filter train independent, the vent on the product filter is left open initially, and the isolation valve to downstream equipment is closed. The product filter vent is closed after air in its housing has been vented. Water is directed to the drains through the barrier filters. Isolating the flow path to the barrier filters can enhance wetting for increased applied pressure drop through a product filter.
Testing: PUPSIT is either a diffusion or bubble-point test (depending on the product filter). In all cases, pressurized gas is applied on the product filter’s upstream side, which is isolated from the system elements both upstream and downstream. Only the drain line with the barrier filters remains open to allow the free flow of test gas through the hydrophobic portion of their membranes.
Drying: Before product is introduced into the filtration line, the product filter typically is blown down and dried to prevent dilution of the product stream. The associated gas is vented through the barrier filter.
Barrier Filter Integrity Testing: Millipak and Millidisk barrier filters are integrity tested offline using 70/30 isopropyl alcohol (IPA) as the wetting fluid.
Process: Once the integrity of the product filter is confirmed, the sterile filtration process can begin.
After Processing: After the sterile filtration process, product recovery through a sterilizing-grade filter can be achieved through air blow-down with application of a low differential pressure (air or nitrogen) of 5 psi to the filter. Users can apply a buffer chase, but product dilution must be accounted for. At the end of product recovery, sterilizing-grade filters are integrity tested with particle-free, water-based, alcohol (70/30 IPA/water), or product-based integrity test specifications. For particle-free–water-based or alcohol integrity-test specifications, a filter must be flushed adequately with the test liquid to remove residual product before testing. Product-based integrity-test specifications can be developed through support from filter vendors.
Design Considerations
Use of Millipak and Millidisk barrier filters in a filtration line is simple and straightforward (Figures 4 and 5). Similar to all critical applications, important process steps and conditions should be reviewed during system and process design to ensure successful implementation of this application. Verification testing should be performed before implementation of an assembly with barrier filters for product filtration.

Figure 6: Sterilization considerations for Millidisk and Millipak barrier filters
Sterilization of Filtration System: Sterilization renders a system or equipment free of microorganisms and is a critical step, especially for aseptic manufacturing processes. Filters can be sterilized through SIP for Millidisk format (cartridge filters) and autoclaving or gamma irradiation for Millipak (disposable capsule) filters (Figure 6). Thermocouples, radiation dosimeters, and biological indicators serve as the worst-case positions within a filtration system and assembly for validation of sterilization.
Flushing and Wetting Product Filters: A filtration system may be flushed and wetted to remove extractables after sterilization as well as for product filter integrity testing. The flushing or wetting liquid passes though the barrier filters to a drain. Flushing/wetting conditions are derived from vendor recommendations (5, 6) and can vary among product filters. Inlet pressures on barrier filters during flushing/wetting procedures should not exceed 0.7 bar. If enhanced wetting of a product filter is required, higher static-hold pressure can be implemented across it. However, the downstream section of that product filter (including the barrier filter) should be isolated during such a high-pressure hold step.
Key Verification Point — Efficacy of Product-Filter Wetting: The efficiency of wetting a product filter(s) through barrier filters can be verified by performing product filter integrity testing. Results can be compared with filter specifications and past trending.
Key Verification Point — Gas Flow Rate of Barrier Filters After Flushing/ Wetting Procedure: Ensuring that the hydrophobic membrane in a barrier filter remains dry is critical. Such dryness can be verified in the filter following a flushing/wetting procedure during the qualification phase with a low-pressure bubble-stream test. This includes disconnecting barrier filters from their assembly and determining their gas flow-rate level at 100 mbar (1.5 psi) pressure, then comparing that to the nominal gas flow-rate level of a new filter unit wetted optimally at low pressure for five minutes. Considering the average hydrophobicity level of polyvinylidene fluoride (PVDF) discs, flow rates in the 30–100% range of nominal rate are characteristic of a “breathing” unit.
Key Verification Point — Using Wetting Medium Apart from Water for Injection (WFI): Compatibility and intrusion pressure/wettability for the hydrophobic filters within barrier filters should be verified before the filters are used. Wetting hydrophobic filters can reduce their air-flow capacity, leading to increased pressure drop across the filter assembly during integrity testing or product-filter blow-down. Millipak and Millidisk barrier filters validation guides provide chemical compatibility information summaries (7–9).
Integrity Testing of Product Filter
Filtration assembly designers should include strategies to minimize product hold by reducing piping or tubing length and to ensure maximum product recovery. The strategy for integrity testing product filters also should be well thought-out, especially for redundant filtration assemblies. In 2012, Felo and coworkers at MilliporeSigma provided an in-depth look at how product filters can be integrity tested in a single-use assembly (10). Their strategy can be applied to stainless steel systems as well.
Interference on Product Integrity Testing from Barrier Filters: Barrier filters are placed downstream of a product filter. To verify the absence of interference, the integrity test result of the product filter both with and without the barrier filters can be compared. Those results should fall within 70 mbar for a bubble-point test and 5% for diffusion flow.
Failure Mode Test: To simulate a worst-case scenario (failure-mode test), users can examine how a fully wet barrier filter gas flow rate compromises a product-filter integrity test. Millipak and Millidisk barrier filters can be fully wetted by flushing with WFI at 3 bar.
Adaptation of Troubleshooting Decision Tree: If a product filter fails its integrity test, users can apply a troubleshooting decision tree such as the example given in PDA’s Technical Report #26 (3).
Drying of Filtration System
To minimize product dilution or contact of product with the wetting liquid (either buffer or water) before filtration, the assembly may be blown down to remove wetting liquid. The current industry practice of blowing down a filtration system ranges from 30 minutes to three hours.
Duration of Drying: Exact drying times should be verified on site and determined during qualification by weight and visual checks. The same time taken to reach the “dry weight” of the assembly will be required for drying the assembly during operation.
Acceptable Applied Pressures: Typical pressures applied for drying filtration assemblies are 0.5 bar higher than the bubble-point pressure of a product filter. Such pressures should not exceed the maximum allowable pressure of the “weakest link” in an assembly. That might be silicone tubing, a connector, or a barrier filter (4.1 bar for Millipak and Millidisk formats), for example. If the required pressure is greater than what the weakest link allows, then blow-down pressure should be reduced, and an extended drying time can be applied.
Absence of Air-Flow Interference: Restriction of air flow through fittings, connectors, tubing, or piping used in an assembly should be minimized. As a product filter dries, the air-flow rate will increase and pressure drop across the product filter will decrease.
Integrity Testing of Barrier Filters
Barrier filters are integrity tested offline with 70/30 IPA/water as a wetting medium and bubble-point test specification of ≥1,280 mbar (18.5 psi). These filters can be wetted by dynamic flushing or static-soak methods. The wetting procedure of barrier filters can be found in a technical guide (5). A 15-minute static soak can be applied to either Millipak or Millidisk barrier filters.
For critical product applications in which resources are readily available, a barrier filter should be integrity tested after the product filter has passed integrity but before product filtration. This minimizes the risk of reprocessing product because of a poor installation or nonintegral barrier filter caused by mishandling. For situations in which resources are limited and product can be reprocessed, barrier filters can be integrity tested after product filtration.
For Best Practices
Barrier filters help enable best practices of aseptic filtration lines for flushing/wetting and preuse integrity testing of product filters. In particular, implementation of Millipak and Millidisk barrier filters is easy and provides flexibility and versatility to the filtration line.
References
1 Annex 1: Manufacture of Sterile Medicinal Products. Volume 4, EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use. European Commission: Brussels, Belgium, November 2008; http://ec.europa.eu/health/files/eudralex/vol-4/2008_11_25_gmp-an1_en.pdf.
2 CBER/CDER/ORA. Sterile Drug Products Produced By Aseptic Processing: Current Good Manufacturing Practice. US Food and Drug Administration: Rockville, MD, September 2004.
3 Technical Report No. 26: Sterilizing Filtration of Liquids. Parenteral Drug Association: Bethesda, MD, 2008.
4 ASTM Standard F838-83: Standard Test Method for Determining Bacterial Retention of Membrane Filters Utilized for Liquid Filtration. American Society for Testing and Materials: Philadelphia, PA, 1983.
5 P35515 Rev G: Wetting Instructions for Filter Units with Durapore Membrane. EMD Millipore: Billerica, MA, April 2012.
6 RF1510EN00: Hydrophilic Durapore Cartridges and Capsules User Guide. EMD Millipore: Billerica, MA, January 2002.
7 VG026 rev 2: Millidisk Cartridge Filter Units with Hydrophilic Durapore Membrane Validation Guide. EMD Millipore: December 1999.
8 VG033 Rev D: Millipak Disposable Filter Units Validation Guide. EMD Millipore: Billerica, MA, May 2012.
9 VG2000EN00: Millidisk Barrier Filter Validation Guide. EMD Millipore: Billerica, MA, May 2002.
10 Felo M, Oulundsen G, Patil R. SingleUse Redundant Filtration. BioPharm Int. 25(4) 2012: 38–41.
Corresponding author Yanglin Mok, BE, is a senior process engineer and technical manager of the Biomanufacturing Sciences Network, 1 Science Park Road, #02-10/11 The Capricorn, Singapore 117528; 65-6403-5313, fax 65-6403-5322; yanglin.mok@merckgroup.com. Lise Besnard, MSc, is a process development scientist at Sanofi Pasteur, 1541 Avenue Marcel Mérieux, 69280 Marcy l’Etoile, France. Terri Love, BSc, is a biomanufacturing engineer; Guillaume Lesage, MSc, is a biosafety technical consultant; and Priyabrata Pattnaik, PhD, is director of the worldwide vaccine initiative; all with the life-science business of Merck KGaA, Darmstadt, Germany, which operates as MilliporeSigma in the United States and Canada. Millipak, Millidisk, and Durapore are registered trademarks of MilliporeSigma.
I read the paper with interest and thank the authors for their efforts. One has to give credit to the hydrophobic/hydrophilic filter combination, as it represents a possible pre-use/ post sterilization integrity test design, a choice of different engineering possibilities. The description of the multiple purposes of the barrier filter are beneficial.
Having said this, I would not necessarily call the described activities as “Best Practices for Critical Sterile Filter Operation”, as anything described promotes the activity of a pre-use/post sterilization integrity test, which, according to risk assessments mentioned in papers, presentations and PDA Position Paper (Vol. 66, No. 5, September–October 2012), represent an elevation of risk. Best practices commonly reduce risks, especially in critical processing steps.
Furthermore, questions arise and clarifications are needed, when one reads the paper.
For example, the statement made that the peruse test will confirm the filter is properly installed, was not damaged during transport and testing after sterilization limits risk. It suggests that all these activities were a) not properly validated and b) the personnel training lacks. In addition, is there a pre-use/post sterilization test performed on the barrier filter, since all of the listed risk criteria are also applicable to the barrier filter ? The authors suggest to perform a barrier filter integrity test, after the product filter passed the pre-use/post sterilization test. The barrier filter test is performed off-line, which means a valve is closed and now is the barrier to my “sterile” filtrate side. The important question would be, how long does one leave the filtration process idle to test the barrier filter ? This time interval to perform the test and leave the filtration process idle requires to be validated, as a downstream manipulation happened. One has to proof that there is no microbial proliferation happening on the upstream side of the tested product filter, which may influence the retentivity. Filter process validations have to take this time interval into considerations and therefore complexity increases.
Drying a sterilizing grade filter, being described as “typical”, means one has to exceed the Bubble Point of the filter and run the drying cycle for a prolonged period at elevated differential pressure conditions, in case of a 0.2 micron PVDF filter >50 psi. How can one be assured that this high differential pressure blow down does not cause damage ? Does one has to integrity test again ? The time and pressure examples are mentioned, but not the necessary validation process to make sure the product filter will withstand the described conditions.
Once again, the paper describes an engineering solution, which can be utilized within a process where the commercial risk is too high not to use a pre-use/post sterilization integrity test. However, this engineering opportunity requires to be as much subjected to a robust risk assessment as a filtration process without the use of a pre-use/post sterilization integrity test.
With regards to the comment from Mr Jornitz, the authors agree that the decision to implement or not implement pre use post sterilization IT should be based on a risk assessment. In the comment, risks associated with implementation were highlighted. However, one should note that there are also risks associated with not implementing a pre-use test, such as product loss due to processing with a damaged filter and regulatory compliance. In many cases, risks of not implementing outweigh the risks of implementation. When implementation is selected, best practices as described in the paper can greatly reduce or eliminate the risks of downstream contamination. In fact, when deciding whether to implement PUPSIT or not, it’s important to consider that a well-designed and operated process, can greatly reduce or eliminate the risk of downstream ingress.
Typically, testing the Barrier filter prior to product filtration might take 30 minutes or less. In any event, feed bioburden must be assessed prior to product filtration. Considering the filter pre-use integrity test is likely done with purified water or WFI and clean dry air, the microbial contamination introduced during the integrity test process should be minimal. If the fluids used for the pre-use integrity test introduces bioburden exceeding validated limits, this may suggest a much bigger problem than the time required to test the Barrier filter.
Furthermore, on the comment on filter drying post integrity test, the drying method described in the article is an effective means of maximizing product yield which is commonly practiced in industry. This practice is routine in many validated processes. For a properly designed, manufactured and operated filter system, air pressure up to the filter manufacturers specification creates no risk to filter integrity.
I have some questions about filtration. Does using autoclave after performing integrity tests, for sterilization purposes, damage the filter or not?
My second question is that filter’s performance is affected by clogging after a period of time, so can I use back wash for removing the clogging or not? how can I find the backwash conditions for a particular filter?
The third question is about performing integrity test and filter sterilization as in-line. How can I find the details of this method?
Thank you very much