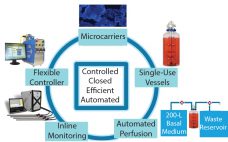
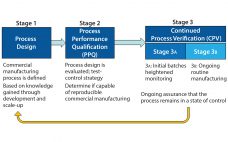
It’s hard to believe that just six years ago, BioProcess International published its first cell therapy supplement, which included just one article on “cell therapy bioprocessing” (1). At the time, most such processing was conducted in special clinical laboratories and academic institutions. As BPI continued to cover this relatively new segment of the biopharmaceutical industry, we heard more about “the product is the process” and “scale out instead of scaling up.” After many trials, errors, and milestones, regenerative medicine has…