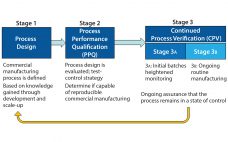
In commercial-scale biopharmaceutical manufacturing, downstream chromatography steps are still a bottleneck and contribute to significant operational costs (1, 2). Some of those costs are inherent (e.g., resins, large buffer quantities, and cleaning) whereas others are avoidable (e.g., product loss due to rejected lots or deviations that result in production downtime). Maintaining efficient and robust chromatography process performance is therefore critical for minimizing operating costs. To do so, we introduce a simple and one-point multiparameter technique (SOP-MPT) for monitoring chromatographic process…