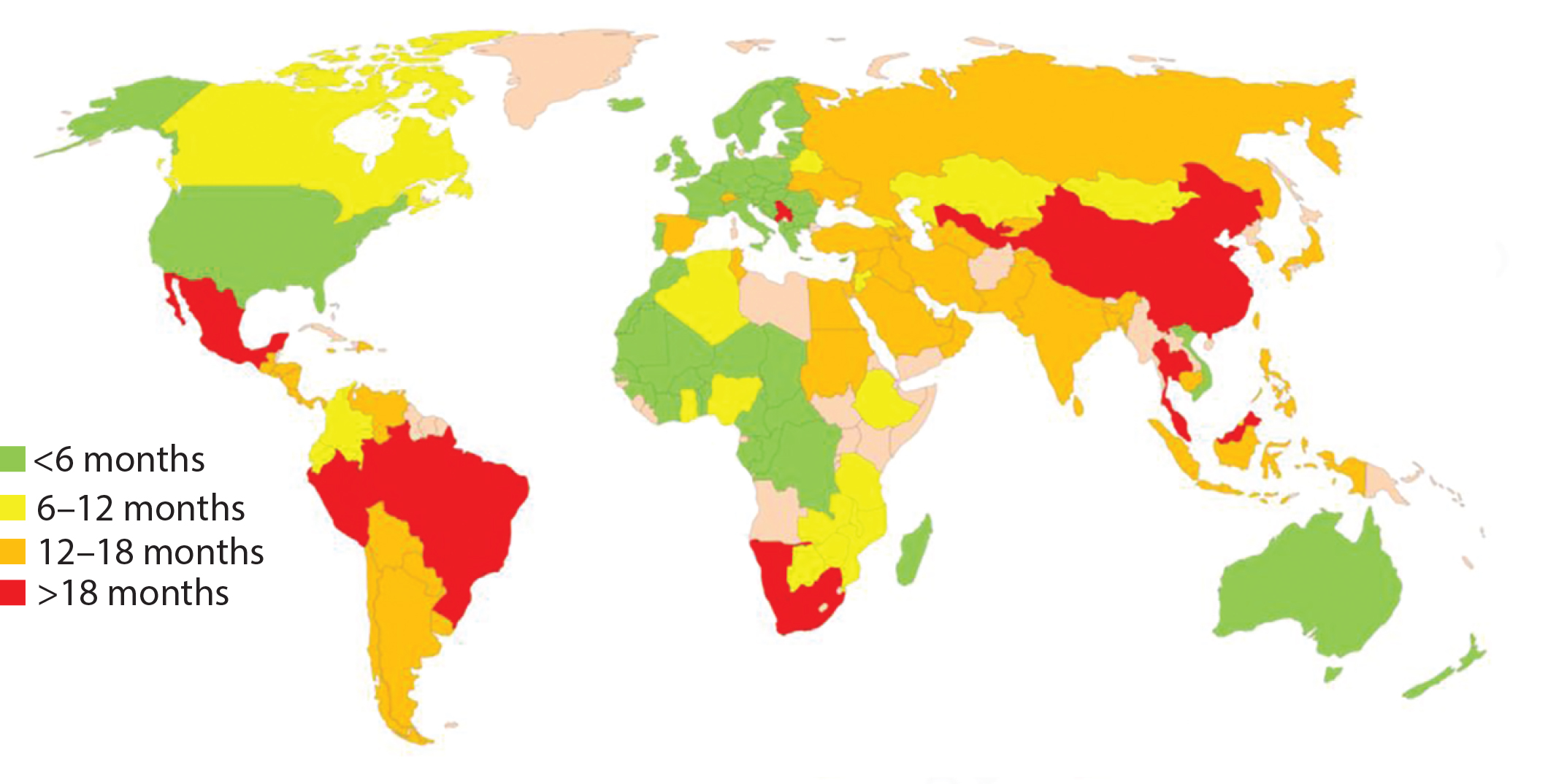
Figure 1: Estimated global approval times for major changes based on industry experience (e.g., new drug product manufacturing site)
Pharmaceutical products save or improve the lives of millions of people each year. Thorough regulatory review of chemistry, manufacturing, and controls (CMC) information is critical to ensure drug product safety, quality, and efficacy as well as to secure patients’ continuous access to such products. But achieving all of that at an effective cost is difficult. Companies race to launch products to patients as soon as possible after clinical efficacy is demonstrated.
Biomanufacturers often need to make changes such as increasing batch sizes, adding new manufacturing facilities to expand patient access, improving analytical methods, and increasing process robustness as companies gain experience in commercial manufacture and testing. Changes also may be made to comply with new regulatory expectations. But before changes are made, manufactures must evaluate process risks and often generate data to confirm that there is no adverse impact to product quality.
As a result of global regulatory requirements, many changes cannot be implemented until health authority review and approval of postapproval submissions. Obtaining approval can take time. The same core data and information can be reviewed by approximately 140 countries, according to local procedures and timelines. Health authorities often have questions during their reviews, which can mean that a manufacturer will need to prepare for many rounds of responses simultaneously. Multiple reviews of the same information, does not improve safety, quality, or efficacy; they can lead to higher costs, a more complex supply chain, and a need for sophisticated systems to maintain regulatory compliance. In turn, that can lead to an increasing number of errors that can affect regulatory compliance, complexity in cost management, and reduced resources as well as increase the risk of an interrupted supply of drug products. Such issues can be more significant for biological products than for small-molecule therapies because biopharmaceuticals have complex manufacturing processes and more stringent regulatory reporting requirements.
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) has implemented reforms targeting wider inclusion of all key regulatory authorities and industry stakeholders to participate in global pharmaceutical harmonization. Following publication of guidelines promoting enhanced product and process understanding, quality risk management, and robust quality systems in ICH Q8–Q11 quality guidelines, the ICH Q12 guideline is being drafted to facilitate management of postapproval CMC changes to promote innovation and continual improvement. The World Health Organization (WHO) issued a guidance document to harmonize health authority approaches to postapproval changes for vaccines (1). Efforts such as these create opportunities for convergence to develop what is needed to enable a scientific and risk-based approach to review postapproval changes so that biomanufacturers can use their global resources wisely and enable fast patient access to high-quality drug products.
Herein we describe the complexities and impact of the current postapproval change management practices, health authority constraints, current mitigation strategies, and recommendations for driving transformational improvements. We aim to communicate a vision of a single, global approval process to benefit patients, health authorities, and industry and encourage the beginning of a journey toward that end.
Complexity and Impact
In about 140 countries, health authorities are charged with the review and approval of documentation to support marketing of pharmaceutical products. Guidelines exist in many of those countries to outline what is needed to support changes made to the products’ CMC. Once changes are submitted, regulatory authorities have greatly varying classifications for those changes in terms of risk to product quality as well as varying timelines for approval (Figure 1). Marketing authorization holders (MAHs, those who wish to market pharmaceutical products) face the following challenges, which can be categorized as regulatory or supply related.
Regulatory-related challenges include
- Understanding and staying abreast of evolving requirements in each country
- Developing and maintaining country-specific versions of similar information to meet individual country requirements
- Maintaining different processes for manufacturing the same product to ensure availability of product made using the approved process for patients in each market, which increases inventory segmentation and potential for errors in terms of manufacturing and regulatory compliance
- Managing inspections that are required for approval of submissions or required before submissions occur
- Support of local retesting by many countries.
Supply-related challenges include
- Designing a supply strategy to cope with varying review timelines and the many different processes approved in each country as a result of various approval timelines
- Delaying implementation of innovative technologies that could increase process robustness or improve analytical methods because of long review times
- Building up sufficient inventory to ensure continuous product supply in markets that are slow to approve changes with no clearly defined approval dates to target
- Delays due to import testing in many countries (2).
Such regulatory and supply problems can lead to increased costs and greater risk of shortages or interruptions in drug-product supply.
Health Authority Constraints
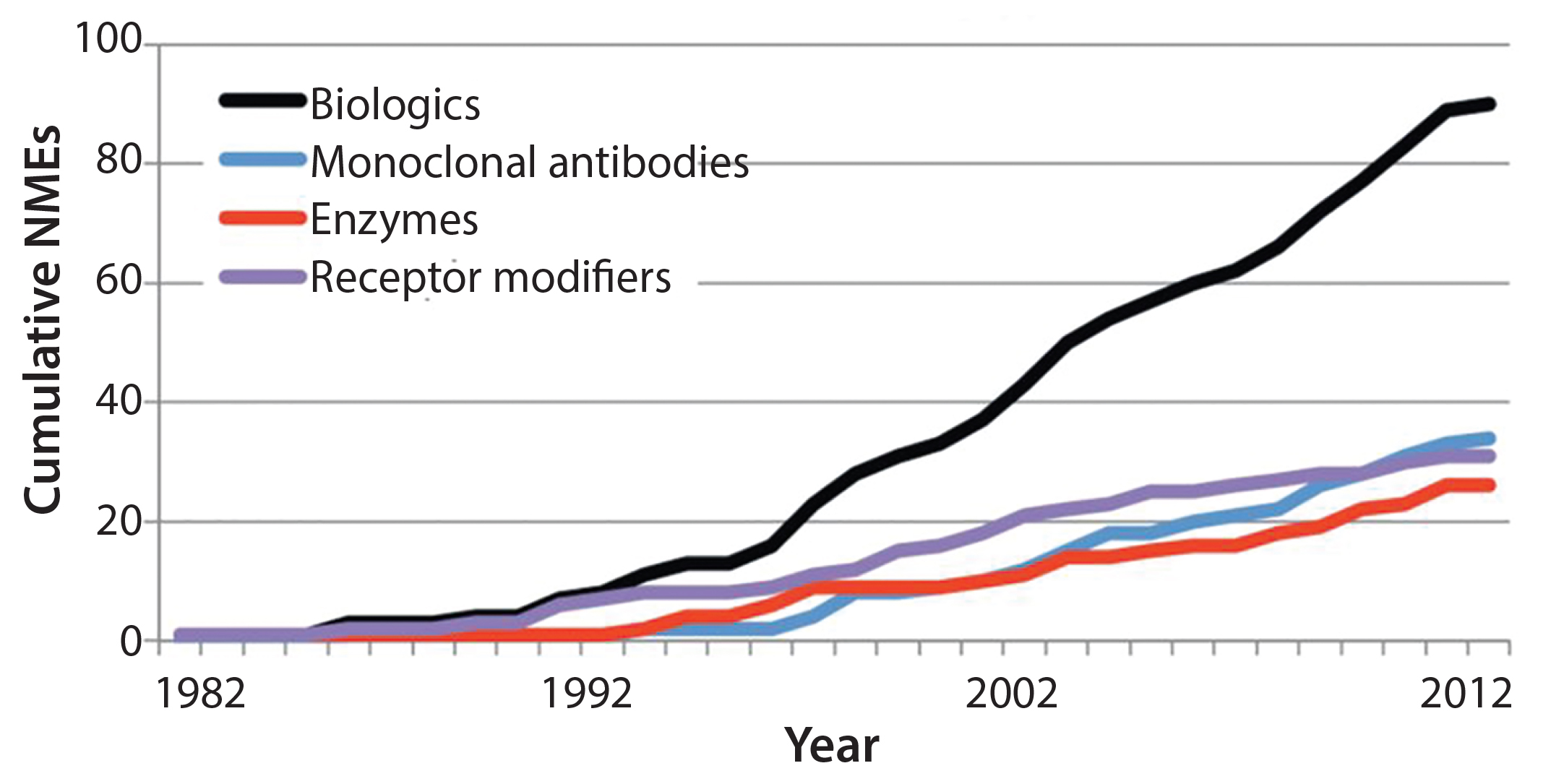
Figure 2: Growing number of new molecular entity (NME) biological products in the United States as an illustration of potential global growth (3)
Health authorities strive to ensure availability of safe, high-quality, and efficacious drug products. However, they faced difficulties of their own when reviewing changes to marketed products. Such problems include
- A need to hire and train enough reviewers in consideration of a growing number of products introduced to the market (Figure 2) and the increasing complexity of those products
- A need for updated guidance or legislation to address issues arising from increasingly complex products and risk-based review based on the significance of the change being proposed, a manufacturer’s product and process understanding, and quality systems in place for manufacture
- Having to address the needs and challenges of regulating manufacturers with a diverse level of product development experience ranging from start-up companies to large, well-established multinational corporations
- Changes in leadership and priorities resulting from government elections, periodic restructuring efforts, and differences in budget allocations.
Mitigation Strategies
MAHs use many strategies to address the above challenges and enable submission and approval of CMC changes, including
- Establishing a database of high-level expectations to include country-specific guidance and historical experience
- Creating templates for CMC information to clearly define detailed expectations based on guidance, historical experience, and agency feedback
- Creating region- or country-specific dossiers to address differences in expectations, reducing the amount of detail in countries that do not require detailed information or do not have laws protecting intellectual property
- Bundling several changes into a single submission to reduce the number of submissions to be reviewed and approved
- Meeting with health authorities, whenever possible, to gain concurrence on strategy before implementation
- Using comparability or postapproval change protocols in markets that permit approval of a manufacturing and/or testing strategy before implementation of a change and a quicker approval once data are available
- Requesting a prioritized review of changes to minimize risks of interruption in product supply.
Health authorities can use their own strategies to address challenges associated with the review of CMC information, including
- Collaboration with other health authorities to promote convergence of policies, harmonization of requirements, and work sharing
- Implementation of postapproval change principles such as those established by WHO in guidelines on procedures and data requirements for changes to approved vaccines (1)
- Implementation of a fee for service for review of CMC changes
- Implementation of an expedited review process that may or may not include a site inspection to allow for faster review of some submissions that meet defined conditions
- Implementation of risk-based reviews in which regulators spend more available time reviewing submissions of higher risk while reviewing and approving submissions of lower risk more quickly
- Attendance at industry workshops to understand the critical elements of product development, manufacturing processes, analytical testing and comparability, technology transfer, scale-up, and postapproval changes
- Reliance on approval of reference country or well-established agencies (e.g., the US Food and Drug Administration and European Medicines Agency).
The strategies used by MAHs can be effective in meeting countryspecific expectations, but significant time, effort, and expertise are needed to customize CMC information for health authorities in each country. Once health authorities receive that information, significant time, effort, and expertise are needed to review it. When balancing the importance of reviewing CMC information to ensure safety, efficacy, and quality and the total resources required on a global scale to approve CMC changes in each market, potential alternate strategies to those listed above emerge.
Ideally, health authorities would define harmonized requirements for approval of CMC changes worldwide. ICH Q12 is being drafted and can help in that regard with recent ICH reforms promoting greater inclusion of global health authorities. The time is now for transformational improvement in management of postapproval changes. Establishment of this guideline could enable MAH to use the principles being proposed in ICH Q12 by expert working group members at industry and health authority conferences, whereby “established conditions” are used as the basis for reducing the number of changes that require health authority approval. Making changes would be based on the applicants’ quality management systems and demonstrated understanding of their products and of which parameters may adversely impact product quality if changed.
Establishment of the guideline also could enable MAHs to use global comparability or postapproval change management protocols for commonly submitted routine major changes (e.g., addition of a new drug substance or drug product manufacturing site) to facilitate quick implementation once data are obtained. Typically, justification for making such a change and the strategy for supporting it are filed with the data and approved worldwide over the months to years that follow. If a comparability or postapproval change protocol was used, the justification and strategy would be filed in the protocol, and the data, if required, would be filed in a separate submission once available. This would be helpful only if health authorities reviewed the protocols within six months and the subsequent data within 30 days.
Additional Advances could be realized through the scenarios below.
Health authorities use the Pharmaceutical Inspection Cooperation Scheme (PIC/S) model of mutual recognition. Participating health authorities follow a uniform set of principles for inspecting manufacturing facilities, and fellow members can choose to approve a facility for manufacture on the basis of another member’s inspection. In the case of postapproval changes, if MAHs follow the principles established in ICH Q12, health authorities could recognize the approval of another health authority that reviewed the postapproval supplement according to those principles.
MAHs could create a global master file for review of CMC information. The information could be posted in a centralized, secure location for simultaneous review by several countries. Reviews could be performed collaboratively during a single time period. Health authorities in each market would have access to the same CMC information (assuming adequate intellectual property protection is in place) and each other’s comments during a defined review period. At the end of the review period, reviewers could meet with the MAH to resolve comments.
Health authorities form consortia to follow a Centralized Authorization Procedure model similar to that established in the European Union. That would enable approval of changes through use of a single marketing authorization by a committee of representatives from all participating countries in other regions.
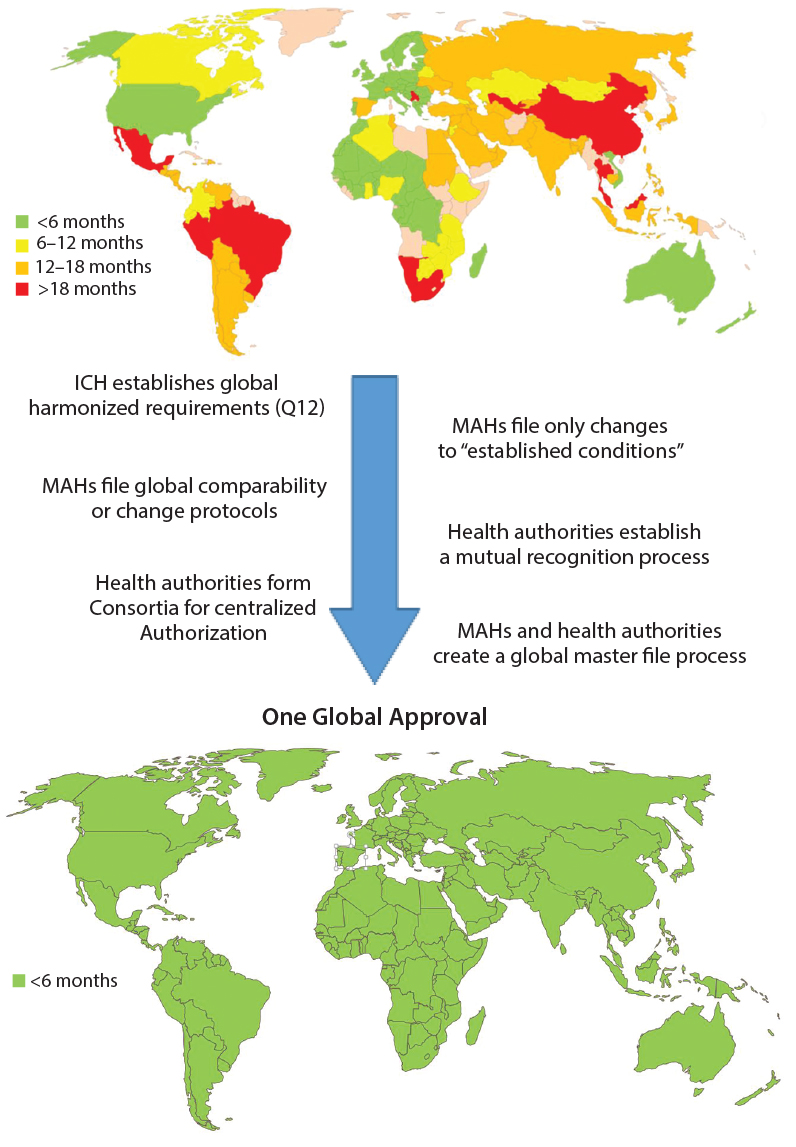
Figure 3: Roadmap toward one global approval (MAHs = marketing authorization holders)
Recommendations
Considering the growing complexity and time and resources needed for getting CMC changes for pharmaceutical products approved globally, it would be in the best interests of patients, health authorities, and MAHs to work together to ensure the greatest possible level of patient access to those products. Although a single, global approval process would provide the quickest solution and require the least global resources with minimal adverse impact on product quality, limitations within health authorities would slow or prevent implementation of such a process now. However, intermediate actions can be taken to journey toward that situation (Figure 3).
Acknowledgments
We thank Bryan Silvey (Baxalta), Tim Wagner (Bristol Myers Squibb), Ann Subashi (Pfizer), Janett Mugaburu-Richards (Pfizer), and Max Fernandez (Baxalta).
References
1 WHO Technical Report Series, Number 993, Annex 4: Guidelines on Procedures and Data Requirements for Changes to Approved Vaccines, Sixty-Fifth Report. WHO Expert Committee on Biological Standardization 2015: 175–259; http://apps.who.int/medicinedocs/en/m/abstract/Js22021en.
2 Appropriate Control Strategies Eliminate the Need for Redundant Testing of Pharmaceutical Products. International Federation of Pharmaceutical Manufacturers and Associations (FPMA) 6 July 2015; https://c.ymcdn.com/sites/casss.site-ym.com/resource/resmgr/WCBP_Speaker_Slides/2013_WCBP_Schreitmueller_Tho.pdf
3 Kinch, MS. An Overview of FDA-Approved Biologics Medicines. Drug Discovery Today 20(4) 2015: 393–398.
For Further Reading
Guidance for Industry: Changes to an Approved NDA or ANDA. FDA, Rockville, MD; www.fda.gov/downloads/drugs/guidancecompliance regulatoryinformation/guidances/ucm077097.pdf.
Guidance for Industry: CMC Postapproval Manufacturing Changes To Be Documented in Annual Reports. FDA, Rockville, MD; www.fda.gov/downloads/Drugs/…/Guidances/UCM217043.pdf.
Guidance for Industry: Established Conditions: Reportable CMC Changes for Approved Drug and Biologic Products. FDA, Rockville, MD; www.fda.gov/downloads/drugs/guidancecomplianceregulatory information/guidances/ucm448638.pdf.
Guidance for Industry: Changes to an Approved NDA or ANDA Questions and Answers. FDA, Rockvillle, MD; www.fda.gov/downloads/drugs/guidancecompliance regulatoryinformation/guidances/ucm122871.pdf.
Comparability Protocols for Human Drugs and Biologics: Chemistry, Manufacturing, and Controls Information (revised draft published April 2016). FDA, Rockville, MD; www.fda.gov/downloads/Drugs/Guidance ComplianceRegulatoryInformation/Guidances/UCM496611.pdf.
New Variations Regulation: Regulatory and Procedural Guidance. EMA, London, England; www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/document_listing/document_ listing_000104.jsp&mid=WC0b01ac0580025b88.
Catherine Hoath is director of global regulatory affairs CMC at Merck Sharp & Dohme Corp. Liuquan (Lucy) Chang, PhD, is associate director of global regulatory CMC biologics at Sanofi. Joseph Kutza, PhD, is director of Regulatory Affairs CMC at AstraZeneca. Kavita Ramalingam Iyer, PhD, is senior specialist, global regulatory affairs CMC at Merck Sharp & Dohme Corp. Heather Smith is director of CMC coordination at Alexion Pharmaceuticals, Inc. Corresponding author Robin Payne, PhD, is at The Studio, Kingfishers Vyne Road, Sherborne St. John, Basingstoke, Hampshire RG24 9HX, UK; 44-771-529-7084; robin@biophorum.com.