A Chemistry, Manufacturing, and Controls (CMC) Strategy Forum was held in January 2012 in San Francisco, CA, to examine the topic of rapid pharmaceutical product development. The purpose of this meeting was to promote an understanding of how best to increase the speed of product development, focusing on areas that improve chances of regulatory success while lessening the time it takes to get a product through development and onto the market. Participants also sought to identify and discuss the issues that accelerate development and those that hold it back — in hopes of developing a winning formula for global best practices.
The concept of “rapid product development” is usually associated with small companies looking to maximize limited resources and achieve a proof of concept that can lead to codevelopment or out-licensing opportunities. The reality is that all companies — small, medium, and large — are looking for opportunities to speed their development to market. Both industry and regulators have the common goal of safely getting life-saving and life-changing drugs to patients in need. But with ever-increasing resource constraints the reality for both parties, significant obstacles stand in the way of rapidly moving products through development, into the clinic, and onto the market.
PRODUCT FOCUS: BIOTHERAPEUTICS
PROCESS FOCUS: MANUFACTURING
WHO SHOULD READ: QA/QC, OPERATIONS, AND PRODUCT MANAGERS
KEYWORDS: RISK MANAGEMENT, FILL AND FINISH, STAFFING, COST MANAGEMENT
LEVEL: INTERMEDIATE
Identified critical-path items were discussed at the meeting with an emphasis on mitigating associated risks to get them “off the critical path and onto the right path.” That would be a way forward through attempts to increase the overall efficiency of a development program by maximizing resources and shortening timelines with the objective of achieving program goals (e.g., benefits for patients through market authorization or out-licensing). Use of modern concepts such as risk management, quality by design (QbD), and prior product knowledge were discussed with emphasis on getting it right the first time. Case studies from industry were presented to review ongoing projects that fit these goals. Regulatory authorities provided comments specific to those programs as well as general guidance and insight on quickly developing high-quality drug products while innovating technologically and ensuring regulatory compliance.
The 22 January 2012 CMC Strategy Forum on Rapid Product Development included case studies presented by both biopharmaceutical companies and regulatory agencies as well as open forums for discussion. We sought consensus on a range of topics related to achieving rapid development of biotech products. The forum consisted of two sessions, each comprising three presentations followed by an interactive discussion with a panel and moderator as well as questions and comments from the audience.

Session One: Strategic Planning
The first session on strategic planning for product development began on Sunday morning, 22 January 2012, with a presentation by Gregg Nyberg of Amgen: “Quality Target Product Profiles and Risk-Based Decision Making.” Nyberg described the use ofmolecular assessments for selecting the most appropriate protein sequence from the start of development. Considerations such as potency, oxidation or deamidation sequences, viscosity, and stability were all considered during selection of the most appropriate protein sequence. Up-front investment in such studies is considered to be an important contribution of QbD for rapid product development because it identifies and “fixes” problems early and thus saves considerable time and money during development of the process and formulation later on. Nyberg went on to cover the uses of product quality risk assessments that allow for prioritization of process development and characterization activities, assessment of changes and mitigation strategies, and control strategy development and refinement. The control strategy justification and rationale are documented in the process.
The CMC Strategy Forum Series
The CMC Strategy Forum series provides a venue for biotechnology and biological product discussion. These meetings focus on relevant chemistry, manufacturing, and controls (CMC) issues throughout the lifecycle of such products and thereby foster collaborative technical and regulatory interaction. The forum committee strives to share information with regulatory agencies to assist them in merging good scientific and regulatory practices. Outcomes of the forum meetings are published in this peer-reviewed journal with the hope that they will help assure that biopharmaceutical products manufactured in a regulated environment will continue to be safe and efficacious. The CMC Strategy Forum is organized by CASSS, an International Separation Science Society (formerly the California Separation Science Society), and is cosponsored by the US Food and Drug Administration (FDA).
The second presentation in this session — “Identifying and Working with Contract Manufacturers” — was presented by Larry Fisher of Roche. Drivers for using contract manufacturing include speed, available capacity, access to technology capabilities, and deferred investment (process or infrastructure) — as well as commercial drivers such as risk mitigation, access to capabilities, and again, deferred investment. Building a network of contract manufacturing organizations (CMOs) that can meet current and future pipeline needs necessitates a gap assessment. Fisher deemed that essential to identifying an appropriate CMO. It would include using risk assessments for contractors (including financial scorecards) as well as risk-mitigation strategies. Once a CMO is chosen, negotiation activities begin. They include a kick-off meeting to develop a plan and organize the tasks and activities required for negotiation. A negotiated supply agreement, term sheet, and quality agreement are all necessary. Then a technology transfer plan is developed that includes detailed project deliverables, a project timeline, a master validation plan, and finally a phase-appropriate CMO governance structure.
The final presentation in the session was “A Regulatory Perspective on the Challenges of Product Development,” presented by Chantal Cazeault, director of Health Canada’s Division of Monoclonal Antibodies. She described the main challenges in product development and pointed out the need to develop a flexible process and control strategy as product knowledge increases. That would help companies meet regulatory requirements at each stage of clinical development. Cazeault stressed that maintaining a link between preclinical and clinical batches, and between those made before and after manufacturing changes as a process evolves are essential components of a successful market authorization application. She also covered several of the most common issues found during the review process that slowed the approval of submissions. Those are addressed in detail below.
Addressing Questions
A
fter a break, the morning presentations were followed by a roundtable discussion of specific questions posed to the presenters and the audience.
How does risk management increase speed and reduce resources? Risk assessments speed development of robust processes through a formal program that helps an organization prioritize efforts and focus resources. Bringing an appropriate level of subject matter expertise into risk assessments forces dialogue between functions that can speed up knowledge transfer and optimize development. A life-cycle approach to risk-informed decision making helps companies assess risks as product and process develop together. Thus can they proactively define the eventual development pathway of their products. Risk analyses also help identify the need for selected use of mitigation strategies throughout a manufacturing process and its development to prevent failures and delays.
Creating common program taxonomy, systems, standards, and training ensures a standardized approach to risk management from early product development through commercial manufacture. Having advanced cross-functional communication and making risk information visible prevents duplication of efforts. The level of product and process analysis and characterization is commensurate with the level of risk, and standardized tools can be used to prioritize mitigation strategies. Assessment of prior knowledge may reduce the need for a company to repeat such experiments, thus saving time and money. Risk assessments can provide a tiered approach to ensure that the most important aspects of development get the most attention. Existing data can be leveraged from assessments already completed. For example, if raw-material risk assessment was completed on Product Family A, then a separate risk assessment for Product Family B may not be required.
Risk assessments performed through the development life cycle help companies identify opportunities for reducing risk through process improvements or redundancies. Theseassessments allow for optimization of product attributes — e.g., critical quality attributes (CQAs) and critical process parameters (CPPs) — for manufacturability, and they ensure that a company measures the right things (e.g., analytical method development focusing on CQAs). Risk assessments can minimize resources through advanced planning — of product runs using design of experiments (DoE) — and provide risk-based controls that reduce analytical development, quality control (QC), and manufacturing requirements, thereby also reducing costs.
Integration of risk management into quality systems provides regulators with greater assurance of a company’s ability to deal with potential risks. It may reduce the extent and level of direct regulatory oversight. This also ensures that quality issues are properly addressed and do not recur (“right first time”), especially if risk-review governance occurs at different stages to ensure that risk is appropriately lowered before a product moves into later stages of development.
From a regulator’s perspective, what are the top five causes of risk and potential delays (e.g., clinical hold)? The multipart answer to this question involves good manufacturing practices (GMPs), comparability, reference standards, potency assays, and regulatory submissions.
Companies need to run their manufacturing plants following GMP to a level that is approvable by an inspectorate. Regardless of how well a manufacturing process is engineered, related assays are developed and executed, risk assessments are performed, and so on — if relevant GMP is not followed, then issues found during a prior approval inspection (PAI) can immediately halt the licensure of a product. Having appropriate quality assurance (QA) oversight is often an issue. A company needs a well-developed and documented quality management system to ensure that product quality decisions and assessments have the correct level of review and sign off.
Often the number-one citation in inspection reports is a lack of appropriately executed investigations for nonconformances. Those can include incomplete product-impact assessments (e.g., not extending findings to other lots), a lack of depth in root-cause analyses, and the need for appropriate corrective and preventive actions (CAPAs).
Product sponsors need to show comparability and understanding of risks following changes implemented throughout development (not making major process changes during late-phase clinical programs). A major regulatory expectation is for companies to show the links in product quality from toxicology studies all the way through a clinical development program to final commercial-scale production. Adequate comparability studies that link changes made to a manufacturing processes during development are essential. Appropriate in-depth characterization of a product — e.g., through lot release, additional characterization, stability, and stress stability studies — is necessary to understanding the effects on any quality attributes. Lacking stability/stress stability data is a common deficiency. How changes in quality attributes can affect safety and/or efficacy also must be considered carefully.
Manufacturers need an appropriately designed reference standard program. Regulatory requirements and expectations are increasing for well-documented reference standards. The need to ensure a link from materials manufactured during development, entered into preclinical and clinical studies, and through to final commercial material is important to prevent a drift in product quality attributes (PQAs). How a reference standard is qualified needs to be justified up front with the same level of rigor as for comparability protocols. A company must justify the standard used in each method and apply appropriate characterization and acceptance criteria for that use. It is essential to prevent drift in product quality, so equal rigor must be paid to the stability, requalification, and replacement of reference standards
An appropriate potency assay is essential. Regulatory agencies consider an appropriate potency assay to be one of the most critical issues for a biological product. Inadequate potency assays can be (and have been) the causes of clinical holds or delays to approval of market applications. A potency assay is needed that, as best as possible, conveys the mechanism of action of a given product. If the product has more than one mechanism of action, then more than one potency assay may be required. Such assays need to be well controlled, reliable, reproducible, and accurate to meet agency and pharmacopeia expectations. (For example, the impact of slope and dynamic range should be assessed.) A life-cycle approach needs to be well planned and agreed upon by the agency — moving from binding-based assays early on to cell-based assays and then possibly back to binding for commercial lot release (if appropriately justified).
Regulatory inspectors say that submissions lack quality through development and at the market application stage. Including an appropriate level of information in a submission helps prevent delays from extensive questioning from a regulatory agency. Presenting data clearly (tables, diagrams, and figures may be helpful) and never assuming that a reviewer knows your program are key. It might be a good idea to have someone not involved in your company’s program review a submission to ensure that the information is clear to an outsider. It is important to include all relevant data to support conclusions so that the agency itself can analyze those data. Including all explanations regarding analyses performed obviates questions. It is essential to ensure that adequate characterization data will be available with appropriate justifications for comparability from lot-release assays, stability, and in-process controls.
What role do process-improvement methodologies (e.g., six sigma and operational excellence) play in speeding up developme
nt? “Lean” is a recurring theme in increasing efficiency, decreasing waste, and using empirical methods for team problem solving (e.g., through Kaizen methods) to decide what matters rather than uncritically accepting preexisting ideas. The focus is on eliminating waste of time, inventory, and movement.
Six sigma is most often associated with improving the quality of process outputs by identifying and removing the causes of defects (errors) using the DMAIC process — define, measure, analyze, improve, and control — and minimizing variability in both manufacturing and business processes. This is distinguished from “lean” approaches in that it uses statistical methods to help identify variability and improvement tools to minimize that variability. Process improvement tools can be useful if applied appropriately by people who are expert in their execution and who also understand the business. They are most useful if applied to a process under development, but they are more often used to improve existing processes. Value-stream mapping evaluates a process to identify bottlenecks and areas of redundancy, thus making it possible to streamline processes and increase their efficiency. A “learning organization” culture allows for lessons learned to be truly woven into existing and future processes so that teams don’t repeate the same mistakes over and over.
Knowledge management is essential to becoming a learning organization. Easy access to readily searchable information is necessary for capturing and leveraging prior knowledge. A focus on human performance leads to more right-first-time outcomes with high-quality products and fast cycle times. Robust qualification programs ensure that staff members have a strong foundation in science, product, and process so that they can respond most appropriately to unexpected events.
What are the advantages and disadvantages of contract manufacturing to increase the speed of product development? Outsourcing can help reduce costs for a product sponsor, both in up-front capital costs and also net expenses because the company pays only for product lots and not the entire cost of a manufacturing plant and its operation. Use of contract manufacturing provides rapid access to technology and equipment that your company may not otherwise have. Contract manufacturers often have broader experience on different molecules from different clients — and should exhibit good GMP compliance due to multiple inspections. CMOs also may be used as a form of risk mitigation if a company wants to have more than one manufacturing site.
In addition, using a CMO can jump-start process development because many such companies work with a range of expression systems and platform processes. Some CMOs offer additional services that can be leveraged by smaller sponsor companies, such as process characterization and validation, analytical development, and regulatory affairs support. Selecting the right CMO, however, requires the appropriate level of due diligence concerning their overall technology, capacity, level of expertise, inspection history, quality systems, staff, and so on. Business and quality agreements must have an appropriate level of detail, considering nonconformances, complaint resolution and timelines, change management and approvals, regulatory inspection coordination, and audit frequency and number of auditors at a minimum.
Forum Cochairs
John Dougherty (Eli Lilly and Company)
Anthony Mire-Sluis (Amgen Inc.)
Program Cochairs
Michelle Frazier-Jessen (Micromet, Inc.–Amgen Inc.)
Shilpa Kaushik (Eli Lilly and Company
Joseph Kutza (MedImmune)
Contracting with a CMO also carries certain disadvantages. First, your company is not the CMO’s only client. The company will have its own quality systems, so you need to understand how your data, standard operating procedures (SOPs), and so on will be integrated into a CMO’s system. You have little control over an outsourcing company’s staff, and it will have its own manufacturing schedules and timelines. Technology transfer also may be more difficult between two companies than within a single company. It can be hard to find a CMO that will provide both the manufacturing and analytical support your company requires. Outsourcing can actually lead to a loss of flexibility because
- Changing the process can be more difficult
- Changes to timing of supply or volume of supply are more difficult
- The manufacturing process usually needs to be finalized sooner with a CMO than with an internal site
- Technology transfer difficulties can be slower to resolve.
Introducing QbD approaches such as process analytical technology (PAT) may be more difficult when your company doesn’t use its own manufacturing plant if it impedes the use of equipment for other CMO clients, creates new processes that complicate the systems in place, or if the CMO is simply not experienced with such approaches. Risk tolerance and tools (e.g., ratings) may not be the same among different companies. So it is beneficial to find a CMO that shares your company’s values (e.g., bringing high-quality medicines to patientsrather than simply focusing on the “bottom line”). Small product-sponsor companies must consider that their product and process knowledge could end up residing at the CMO if all CMC work is performed there. So it is very important that a sponsor determine how it will internalize knowledge being generated by a CMO.
Session Two: Speeding Drug Development
The theme of the next session was how to apply better science to reduce drug development time. The afternoon began with a presentation by Sonya Schermann of Micromet (now Amgen): “Using Cell Culture Samples to Provide Early Indicators of Product Quality.” She described how biotechnology products are typically produced in cell culture and followed by a multistep purification process. Partial purification is usually required before analytical characterization of a product. That approach is time- and resource-consuming, and analysis of purified product may not accurately reflect a molecule’s attributes in a cell culture supernatant or harvest sample.
Micromet developed a technique that allows product characterization at the earliest stages of the manufacturing process, including cell line and upstream process development. This technique involves measuring product mass using electrospray ionization mass spectronomy (ESI-MS), which allows for direct analysis of cell culture supernatant. The result is a high-throughput screening of fermentation conditions, for example, in samples from a 96-well plate. Mass spectrometry can be used to monitor the mass of intact proteins or to monitor the mass of peptides produced by a proteolytic digest. Intact protein measurements can be used to detect any modification that produces a mass change of about >10 Da. Peptide measurements can be used to localize modifications and to detect modifications that produce mass changes of ≥1 Da (e.g., deamidation, disulfide bond formation). This technique requires a relatively simple sample preparation that mitigates the risk of altering variant composition during purification. So it is a faster, more accurate examination of variants at every stage of manufacturing process development than would be possible with other currently available analytical techniques. The method improves companies’ ability to design an effective, efficient manufacturing process at all stages.
The Arrhenius Equation
The Arrhenius equation is a simple formula expressing the rate of a chemical reaction. It was first proposed by Svante Arrhenius in 1884. Seen as an empirical relationship, the equation can be used to model temperature variance of diffusion coefficients, population of crystal vacancies, creep rates, and many other thermally induced processes and reactions. A useful generalization
supported by the Arrhenius equation is that, for many common chemical reactions at room temperature, the reaction rate doubles for every 10 °C increase in temperature.
The Arrhenius equation gives “the dependence of the rate constant k of chemical reactions on the temperature T (in absolute degrees Kelvin) and activation energy, Ea,” as shown below:
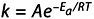
where A is the preexponential factor (prefactor) and R is the universal gas constant.
The second talk in this session was “A Risk Based Strategy for Manufacturing First-in-Human Enabling Toxicology Study Material,” by Tongtong Wang of Eli Lilly and Company. Wang described an approach to shortening cycle times from candidate selection to producing material for nonclinical trials. Essentially, the program moves away from a traditional selection of the top four clones after transfection followed by selecting the top candidate to produce a toxicology lot. In Lilly’s innovative process, material is made from a pool of the four top clone candidates. That can reduce the time needed for early development by about 4.5 months. Pooling clones is assessed through a risk-based approach that depends on characteristics of the molecule under consideration (e.g., glycosylation differences among clones would not change effector function of an IgG4 molecule). Assessment of charge variants is often qualitatively the same, with typical changes in distribution of profiles rather than new peaks being present.
Most charge variants pose no safety concerns. However, it is possible (although rare) that genetic changes might be detected by analytical liquid chromatography and mass spectrometry (LC-MS), although their presence in toxicology material is unlikely to cause a toxicological event. The risk is mitigated through structural characterization of the top four clones. Therefore, the risk is low that clone-dependent quality attributes that could affect toxicology studies, especially for IgG4 molecules and when characterization of the top four clones is carried out before pooling. This strategy removes production of nonclinical material off the critical path and onto the right path at the expense of it being more complex to scale up four clones at a time instead of one.
The next talk was “Applicability of Arrhenius Modeling to Protein Products: Reducing the ‘Wait’ Time for Stability Data to Support Expiry Dating,” by Brent Kendrick of Amgen. He described how waiting for stability data can be a critical-path item on the investigational new drug (IND) and market authorization application (MAA) submission timelines. Setting expiry limits (typically two to three years) requires knowledge of the degradation mode and rate. Two options exist for determining the extent of degradation:
- Placing actual clinical or commercial-scale material on stability and monitoring over the full time course
- Placing actual clinical or commercial-scale material on stability and monitoring over part of that time and extrapolating based on representative historical material stability kinetic profiles and/or using Arrhenius modeling for those degradation pathways shown to follow Arrhenius behavior.
Detailed in the box on the previous page, the Arrhenius Equation describes how reaction rates depend ontemperature and activation energy. The goal for application of Arrhenius kinetics in protein stability and shelf-life determination is to extrapolate the rate constant down to a recommended storage condition (2–8 °C). In many cases, initial degradation rates fit to a linear model when the extent of degradation is low — for example, at early time points in accelerated conditions. The results are often valid out to many years in recommended storage. However, Arrhenius extrapolation to higher temperatures may underestimate the degradation rate (especially as those temperatures approach the onset of unfolding melting temperature, Tm). So in some cases Arrhenius extrapolation is valid only across narrow temperature ranges (~▵T = 10 °C). Thus, there are different options to extend expiry: Arrhenius methods and others.
Some degradation pathways do follow Arrhenius kinetics. If they can be established early on for a product, you can use accelerated data (usually limited to Tm is approached), protein structure becomes destabilized, which often accelerates physical and some chemical reactions beyond Arrhenius predictions. That may affect extrapolation to lower temperatures.
Non-Arrhenius extrapolation of limited time-point data at recommended storage for expiry on clinical or commercial material may be justified. This works if stability data are available on representative material that fits a robust model, and if sufficient data at recommended storage (one year minimum) exist to provide sufficient time points for accurate extrapolation — and elevated temperature and stressed stability profiles are comparable. We recommend discussion with regulatory authorities before using Arrhenius modeling to predict shelf-life.
The last presentation of the session was “Issues That Impact Product Development: A Regulatory Perspective,” by Barbara Rellahan of FDA/CDER’s Division of Monoclonal Antibodies. She described top issues that the FDA finds to affect timing of product development: underestimating the risk associated with and/or data needed to support a manufacturing change (e.g., process changes, assay replacement, method alteration, site change); underestimating the risk or insufficiency of data to support “new technology”; poor-quality management and/or quality control systems; and not following advice given in pre-IND meetings.
For example, risk can be underestimated when a company doesn’t know how much stability data are required to support a change when that change could affect product stability. Companies underestimate the impact that a method change could have on method performance, the amount of data needed to support replacing one assay with another, as well as the risk that a change will affect a CQA. Such an impact can be compounded by poor understanding of a product’s CQAs. The resulting data may be insufficient to support a change, leading to a product hold, especially if there are safety concerns (e.g., if the product mediates cytokine release, or if other severe toxicity is associated with it).
Rellahan recommended using risk assessments to develop a preliminary ranking of quality attributes by their criticality as early in development as possible. Before phase 1 and during early development, such assessments help companies prioritize and focus early characterization efforts. Before making a manufacturing process change, companies can assess risk associated with a change and focus their comparability work. She also recommended that sponsors listen to advice given during pre-IND and IND meetings. Not following such advice could lead to delays in product development. It is also important to invest in a pharmaceutical quality system (1). That helps enable efficient knowledge and quality risk management as well as continual improvement efforts.
Roundtable Discussion
Following the presentations, a roundtable discussion with the presenters and the audience addressed a number of questions.
From an industry perspective, what top critical-path activities tend to
limit time to approval? Preclinical and clinical trials take an extensive period to complete, but CMC work is not usually on critical path for MAAs. Getting enough stability data — for both comparability studies and determining an acceptable shelf-life — can certainly limit time to approval. So can validation (process, method, shipping, and so on), process development, technology transfer, analytical method development (especially for bioassays), product characterization reference standards, and product characterization. Writing and receiving single-cycle review and approval of regulatory submissions are time-consuming activities, as is scheduling with CMOs.
What technologies or approaches can provide incremental increases in the speed of product development? The answer to this question is complex and involves biomarkers, process development timelines, single-cycle development, analytics and automation, technology transfer, product characterization, and interactions with regulators.
Develop better biomarkers for clinical end points. Timelines for clinical studies often depend on the end points being sought. Examples include overall survival, tumor shrinkage, and reduction in joint damage. They can be precluded with good biomarkers that extrapolate to efficacy or safety. Selecting appropriate clinical end points can make the difference between success and failure in a clinical trial.
A company must have a well-planned stability program to ensure product quality, meet regulatory expectations, and prevent unnecessary delays. This should include accelerated/stress studies that can reduce real time requirements, use Arrhenius modeling when applicable, and have appropriate lead and supporting lots. Companies should leverage formulation development data that can increase expiry dating beyond final commercial-scale material. Andmaterial should be used from early representative runs (e.g., engineering or pilot-scale confirmation runs) to establish lead lot stability.
According to a validation life-cycle approach (2), using development data may reduce the need for the rule-of-thumb three conformance runs. Companies may be able to spread validation work out to include continuous monitoring, but this life-cycle approach has yet to be accepted by all regulatory authorities.
Process development timeliness can be shortened. Manufacturability assessments focusing on a target product profile are of great benefit if the correct protein sequence and clone are selected early in development. They can reduce manufacturing process requirements (because of fewer variants, higher yields, an absence of aggregation, better stability, and so on). Strategies that involve pooling clones can be used early in development to create a toxicology lot without having to wait for final clone selection. That can allow more variability to enter preclinical studies, which may in turn help companies develop CQAs and product specifications.
Single-use manufacturing technology was also discussed as a way to speed development. But related concerns must be considered such as the need to assess the effects of extractables and leachables on both cells and products. The main advantage of disposables seems to come with fewer requirements associated with cleaning procedures and cleaning validation.
Single-cycle development. Single-cycle approaches to development are preferable. The more that products or processes are changed later in development, the greater the risk to their successful approval. Leveraging clinical lots to provide material for process characterization studies eliminates the need to complete manufacturing runs specifically to generate material for such studies. Running clinical batches at a commercial facility allows comparability and validation runs to be performed at same time, reducing the number of engineering runs required and the timing between those runs and the validation campaign. Companies can manage risk at commercial scale using experience gained over past technology transfers to identify specific testing required before running product batches. Process characterization experimentation can be overlapped with report writing to streamline documentation efforts.
Automation. Automated data gathering (for analytical methods, in-process testing, manufacturing equipment parameters, and so on) with data storage (e.g., manufacturing data warehouses and electronic laboratory notebooks) can greatly speed up drug development, as can data analysis and searchability.
Enhanced technology transfer. Companies often move their manufacturing processes from pilot/clinical facilities to scaled-up commercial facilities. Associated technology transfer times can be improved through value-stream process mapping to streamline the overall process. Using small and pilot-scale studies to reduce engineering runs through a risk-based approach saves time and money overall. By establishing and implementing platform processes and methods, a company can leverage information and experience from previous technology transfers to speed up subsequent ones. That can lessen requirements for equipment and facility modifications as well as for new raw-material orders.
It also creates less burden from novel method transfers, and so on.
Analytics of the future could offer more rapid product analysis using mass spectrometry. Direct analysis of fermentation supernatants and peptides by ESI-MS provides powerful resolution and variant identification. Purification-free Lys C nanoinfusion MS and reduced size-exclusion MS methods shorten analysis times for glycoforms from eight hours (with familiar chromatographic methods) to 30 minutes. You can also speed up high-performance liquid chromatographic (HPLC) methods using ultraperformance liquid chromatography (UPLC).
Analytical platforming. Putting well-known methods that are not product-specific into practice early in development is especially useful for monoclonal antibodies (MAbs), for which most quality attributes are already known because they are similar for all antibodies. This reduces the time needed for early method development and allows for templating regulatory submissions and quality documents. And that can reduce the time needed for method transfer and subsequent development.
Enhanced product characterization methods improve comparability and prevent unknown quality/safety/efficacy issues. More sensitive and accurate analytical methods are constantly under development — such as for subvisible particles (e.g., NanoSight, microflow imaging, and HIAC particle counting), MS, improved capillary methods, and more sensitive tertiary structure methods (e.g., nuclear magnetic resonance and Raman spectroscopy). However, caution should be taken when implementing such methods because more sensitive analytical methods could bring an increased regulatory burden to identify and control smaller and smaller levels of variants. We should be tuning our methods to focus on CQAs rather than simply the number of peaks on a chromatogram.
More rapid submission writing and single-cycle regulatory review. One of the best approaches to speeding drug development is to interact withregulators. Joint collaboration on critical items (don’t waste regulators’ time) improves your chance that submissions will proceed without issues during development. Listen to regulatory feedback both from the agencies and your own regulatory affairs department and include that in your submissions. Another useful approach is for sections of regulatory documents to be written as data and knowledge are generated.
How can QbD help speed up product development, and where does investment in time and resources provide the most return? The target product profile provides end points to aim for that can lead to design of a manufacturing process for creating a product as intended. Manufacturability assessments can provide the biggest return for up-front investment in time. Sequence and
hot-spot analysis can be used to engineer a product to CQAs and to fit to a first-in-human (FIH) platform. High productivity can be selected with optimal signal peptide use. Protein compatibility can be selected with the FIH platform formulation based on the ability of a molecule to be stable in serum and process buffers, determined through pH and serum-based jump studies.
Particulation propensity can be screened with predictive particulation assays. Companies can develop degradation-rate databases and Arrhenius models to enable decisions based on the rate predicted for 2–8 °C. Concentration and viscosity screens are valuable as well, making it possible to engineer-in low-viscosity attributes.
As described, risk assessments provide a way to improve cross-functional collaboration and information exchange. They allow companies to focus their resources on the most important issues and predict risk concerns up front, then fix them before problems occur. Leveraging prior knowledge can save a great deal of time and resources by preventing companies from “reinventing the wheel.” However, be cautious in interpreting data from another molecule to your own because biotechnology products inherently surprise us.
Permanent Advisory Committee for These Forums
Siddharth Advant (ImClone)
John Dougherty (Eli Lilly and Company)
Christopher Joneckis (CBER, FDA)
Rohin Mhatre (Biogen Idec Inc.)
Anthony Mire-Sluis (Amgen, Inc.)
Wassim Nashabeh (Genentech, a Member of the Roche Group)
Anthony Ridgway (Health Canada)
Nadine Ritter (Biologics Consulting Group, Inc.)
Mark Schenerman (MedImmune)
Keith Webber (CDER, FDA)
Using QbD principles, a company can gain understanding of CQAs and CPPs, which leads to process development aligning with the criticality of product attributes, more focused and rapid process characterization, and a streamlined development of the control strategy. Design space and multivariate studies may not be of much value for speed to development, but they can provide extensive savings and opportunities after market approval.
What approaches can smaller companies use to speed their drugs to market? The virtual company is one obvious strategy: outsourcing work to contract manufacturers and contract laboratories and using clinical research organizations for preclinical studies and clinical trials.
Although consultants can be helpful, it’s also important for small companies to employ some experts of their own: people who are vested in the interests of the company and in turn will drive its success. Using risk assessments to drive what is necessary over what is nice to have is an important exercise. It is important to have staff or consultants who are knowledgeable in facilitating risk assessments. Such exercises require no capital cost and can lead to significant increases in efficiency and lowering of costs over the long term.
Careful planning of resources should help a company meet specified goals. Consider what to front-load before seeking partners or extra funding, and determine how to decide on outcomes before driving next steps. Small companies can also benefit by leveraging prior and public knowledge as well as interactions with regulators and partnerships with bigger companies.
Author Details
Corresponding author Anthony Mire-Sluis, PhD, is North American vice president of contract and product quality at Amgen Inc., MS-37-2-C, One Amgen Center Drive, Thousand Oaks, CA 91320-1799; 1-805-313-2415; anthony.mire-sluis@amgen.com. Joseph Kutza, PhD, is director of CMC global regulatory affairs at MedImmune. And Michelle Frazier-Jessen, PhD, is director of CMC regulatory affairs at Amgen Inc.
REFERENCES