 This two-day CASSS CMC Strategy Forum explored many technical, practical, and regulatory facets of biological drug-product (DP) analytics, process validation, and comparability. Part 1 of this report summarized the discussions on drug-product analytics and comparability in BPI’s March 2021 issue (1). Here we report on day two presentations and discussions on validation, legacy products, and lifecycle management.
This two-day CASSS CMC Strategy Forum explored many technical, practical, and regulatory facets of biological drug-product (DP) analytics, process validation, and comparability. Part 1 of this report summarized the discussions on drug-product analytics and comparability in BPI’s March 2021 issue (1). Here we report on day two presentations and discussions on validation, legacy products, and lifecycle management.
Session Three: Drug-Product Validation
The morning session focused on principles of process validation with examples of challenges specific to drug products.
New Risk-Based Process Performance-Qualification Strategies: Patrick Donahue of Janssen began by reviewing the US Food and Drug Administration’s (FDA’s) guidance on process validation (2, 3). It introduced the concept of a control strategy driven by relative risk and mitigation that is commensurate to the type and level of risk.
Criticality of an item (e.g., parameter, process, or equipment) is the guidepost for monitoring and control: The higher the criticality, the more controls are placed on a parameter, and the tighter its range will be. Criticality arises from a parameter’s impact on product quality and uncertainty about the process–product relationship. Because uncertainty is high in the early stages of a program, criticality can be high initially and decrease as more process understanding is gained over time. Impact usually is based on science and prior knowledge. In graphics adapted from Ken Hinds’s paper (4), Donahue depicted a control strategy based on either sensitivity of a given parameter to affect product quality or sensitivity of a quality attribute to affect clinical safety/efficacy — and the latter enables setting of clinically relevant specifications.
Donahue described three stages of validation (Figure 1):
- process design or process development stage, in which understanding is gained regarding the source of variations, their effects, and how to control them
- process performance qualification (PPQ) stage, in which commercial facility, equipment, and utilities are established and data are used from scale-up and scale-down studies with enhanced sampling to demonstrate that a process can withstand variation and meet specifications; setting statistical limits at this stage may be difficult because all parameters might not have been explored yet
- continuous process verification (CPV), in which enhanced sampling continues to ensure that a process remains in a state of control.
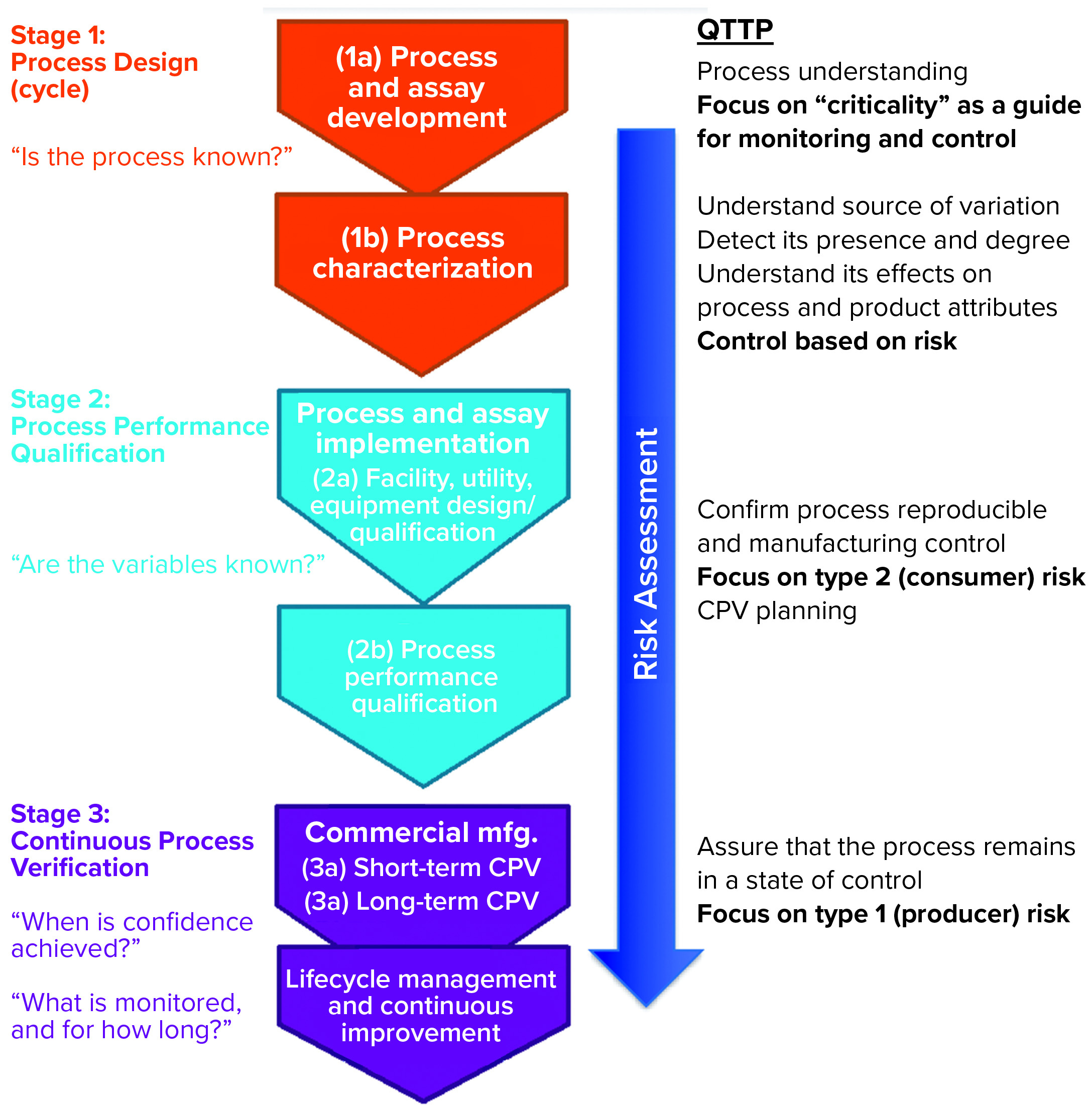
Figure 1: Three stages of process validation; QTTP = quality target product profile
In the PPQ stage, the focus is on controlling the type 2 (beta) risk of releasing a lot that has failed to meet an attribute standard; in the CPV stage, companies must manage the type 1 (alpha) risk of failing a good batch.
Donahue emphasized the importance of sampling strategy to help process engineers understand the sources of variation. He recommended taking many samples throughout development. Because the number of drug-product lots often is limited in early development, sponsors should try to understand variability within a given batch. For example, a standardized approach to assessing drug-product homogeneity (within batch variability) is to sample from the beginning (the first 10% of manufactured units), middle (mid 20%), and end (last 10%) of a batch.
Donahue described some statistical tools used in stage 2 (PPQ in Figure 1), including tolerance intervals (TIs) to measure within-batch homogeneity, analysis of variance (ANOVA) to measure both within-batch homogeneity and between-batch consistency, and partitioning of variability into variance components. He emphasized that data should not be rounded before analysis and cautioned that statistical significance is not always the same as practical significance.
In stage 3 (CPV, Figure 1), developers still are gaining knowledge about variation from raw-material attributes and process parameters, so extended sampling should continue. Donahue presented a statistical approach to using attribute severity and adjusting the width of TIs to determine when enhanced sampling can be stopped. He ended his talk by specifying that good statistical process validation improves a company’s ability to detect problems, find their root causes, improve product quality, and decrease patient risk.
| CMC Strategy Forum July 2016 Scientific Organizing Committee |
| Siddharth Advant (GlaxoSmithKline, previously with Celgene Corporation), David Allen (Eli Lilly and Company), Natalya Ananyeva (CBER, FDA), Howard Anderson (posthumous, formerly with CDER, FDA), Yves Aubin (Health Canada), Julia O’Neill (Moderna Therapeutics, formerly with Tunnell Consulting), Zahra Shahrokh (STC Biologics/ZDev Consulting), and Andrew Weiskopf (Sana Biotechnology, formerly with Biogen) |
Process Validation of a Drug Product with Multiple Stock Keeping Unit (SKU) Numbers: Kevin Maloney of Biogen presented a case study using science and prior knowledge to manage the complexity of multiple concentrations, fill volumes, DP sites, and drug-substance (DS) sites. Referring to the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), Biogen conducted PPQs in DP launch line 1 for ICH regions, and changes to the DP fill line occurred post-PPQ for markets in the rest of the world (RoW).
The stage 1 strategy (Figure 1) was to use a single, fully formulated DS at the highest protein concentration with different fill volumes because concentration rather than fill volume drove stability. This approach simplified future validation and bracketing stability strategies. For stage 2, facility designs were similar for commercial line and prelaunch lines, and a platform fill–finish process was used, so the two lines were considered to be equivalent. A bracketing approach of three fills under extreme conditions was based on surface:volume ratio, headspace:volume ratio, and the ratio of stopper area to volume. In addition, the PV included one fill of each intermediate dose. Maloney mentioned that a risk for bracketing in stage 2 is that the dose is not known yet, so the company will file for licensure based on the entire bracket.
Drug Product Sterility Assurance: Colleen Thomas of FDA’s Center for Drug Evaluation and Research (CDER) presented an FDA reviewer’s perspective on sterile DP process validation, referring to laws and regulations governing sterile products (5–8). Regarding sterilizing filtration, reviewers expect to see information on integrity testing, process parameters, microbial retention, postreconstitution storage, postdilution storage, and media fills (9).
Thomas provided several examples of deficiencies in recent biologics license application (BLA) submissions. In filter-integrity testing, for example, one sponsor provided no information on product bubble-point determination; test descriptions were insufficient and used only a “pass” acceptance criterion rather than providing numerical values; and a filtration time limit was not provided (or was significantly longer than that validated by microbial retention studies). She further indicated that for microbial-retention validation, information gained from comparing scaled-down study and production parameters must be provided along with a retention-study report and viability data. One company’s deficiency was its inadequate justification for a microbial-retention study that used just water; the sponsor was asked to repeat the study with a suitable surrogate to model DP attributes.
For lyophilized products, Thomas indicated that postreconstitution storage times must be supported by microbial challenge studies using the panel of microorganisms listed in chapter <51> of the US Pharmacopeia on antimicrobial effectiveness testing as well as typical skin flora or species associated with hospital-borne infections. For in-use studies, the recommendation is to study two storage conditions (because hospital refrigerators might not be well controlled). In one case, a sponsor had proposed up to 12 hours for ambient storage of diluted DP, but the growth-promotion property of the diluted product did not support ambient storage. Thomas provided examples of media-fill deficiencies:
- lacking descriptions of media-fill relevance and manufacturing processes
- insufficient media fill data
- unvalidated maximum hold times
- nonexistent action plans for media failures.
Lyophilization Validation: In the final talk of the morning session, Ellen Huang of FDA’s Center for Biologics Evaluation and Research (CBER) offered considerations for validation of freeze-dried DPs, starting by listing several examples of deficiencies. She described the key elements of lyophilizer equipment qualification, which include empty-chamber temperature mapping, leak-rate testing, and condenser capacity measurement.
In stage 1 (PD in Figure 1), a developer needs to understand the critical properties of its DP formulation: thermal characteristics, stability, and excipient properties. Laboratory, pilot, and at-scale runs demonstrate the effects of parameters such as shelf temperature, chamber pressure, time, and ramp rate to define a design space. In stage 2, a sponsor demonstrates its ability to manufacture uniform products consistently with temperature mapping and extended sampling. Boundary studies are conducted using shelf temperature and chamber pressure offset from their commercial setpoints.
Typically, three runs are performed at maximum load and one run at minimum load. Bracketing can apply to load, lyophilizer, dosage-strength, fill-volume, and vial-size specifications. Huang discussed elements of validation for cleaning, sterilization, and aseptic processing, which for lyophilized products include media-challenge studies covering transportation from fill line to lyophilizer along with loading, holding, partial vacuum, stoppering, and unloading. In stage 3, trend analysis of data and sampling/monitoring might be decreased once sufficient knowledge is gained on the impact of material lots over time.
Huang also discussed some special cases such as orphan therapeutics, tray-lyophilized drugs, and syringes prefilled with freeze-dried product. For orphan drugs with little material available for testing, sponsors can use appropriate surrogates, perform seeded PPQ runs, or submit fewer PPQ runs and perform additional runs in the future to support process performance qualifications.
| CMC Issues |
| The CMC Strategy Forum series provides a venue for biotechnology and biological product discussion. These meetings focus on relevant chemistry, manufacturing, and controls (CMC) issues throughout the lifecycle of such products and thereby foster collaborative technical and regulatory interaction. The Forum strives to share information with regulatory agencies to assist them in merging good scientific and regulatory practices. Outcomes of the Forum meetings are published in this peer-reviewed journal to help assure that biopharmaceutical products manufactured in a regulated environment will continue to be safe and efficacious. The CMC Strategy Forum is organized by CASSS–Sharing Science Solutions and is supported by the US Food and Drug Administration (FDA). |
Panel Discussion on Process Validation
For the panel discussion on drug product comparability, speakers and cochairs from the first session were joined by Sara Gagneten of CBER.
Demonstrating Comparability of a Second Fill Line: Kevin Maloney responded to a question about the types of data generated to claim that a second fill line is comparable to the first one. He indicated that the elements of risk assessment and quantitative scoring should be applied to both lines — with stability data generated from the second.
Demonstrating Microbiological Safety with In-Use Studies: One question asked of Colleen Thomas regarded the rationale for recent expectations about conducting microbial challenge evaluations as part of in-use studies. Her response was that many biologic products support microbial growth, so regulators want to know what the risk to patients will be. Label adjustments may be appropriate. Generally, four hours at 2–8 °C is acceptable for in-use studies without data, but if longer times or ambient storage are claimed in labeling, then supportive microbiological data should be included along with biochemical stability data. A BLA would be expected to include both types of data, which might be asked about during an end-of-phase-2 meeting.
Transition from Stage 3a to Stage 3b of Process Validation: Patrick Donahue responded to a question about timing the transition from stage 3a to 3b (Figure 1). He said that it occurs typically after 25 batches or one year into stage 3a. When asked about the number of assays used to test enhanced samples, he said that usually two or three of 15 release assays will be added to enhanced sampling. A protocol would be written to evaluate the results of the first five batches during stage 3b for determining whether enhanced sampling still is required.
Criticality, Uncertainty, and Severity: Stefanie Pluschkell of Pfizer brought up a concern about the “criticality = impact × uncertainty” rule of thumb. She pointed out that there could be times when uncertainty is low but severity of an adverse impact is high, thus making the criticality high as well.
Formulation Robustness Studies: Patrick Donahue responded to questions about the timing and design for formulation robustness studies. He said that more rigorous and systematic formulation robustness studies often are performed in late phase 2 and/or early phase 3 clinical programs based on which control over excipient ranges would be justified.
Process Validation for Special Cases: Julia O’Neill of Moderna Therapeutics commented that, in her experience, the lines between stages 1 and 3 (Figure 1) in PPQ are blurred for breakthrough therapies, as they are for orphan drugs.
When sponsors submit BLAs that include data from only one PPQ lot, the FDA expects them to initiate a type C (end of phase 2) meeting to describe the follow-up plan. Clinical lot data sometimes can be leveraged, but only if they are collected under protocols using prospective criteria and with the same rigor as data collected for process validation. In one example of a breakthrough orphan drug, a company submitted data from one PPQ lot using a template process in its BLA, leveraging all development and clinical lot data along with a commitment to extensive monitoring throughout the lifetime of the product.
A comment was made about in-use study strategy for a biosimilar product. The originator had claimed that diluted product for administration was stable for 24 hours when refrigerated. The questioner pointed out that FDA expectations for in-use–study hold times have changed since the originator product was approved and thus might require modifying the biosimilar label based on biochemical and microbiological data.
CPV or Comparability Protocol: Regulators are finding more comparability protocols than CPV plans in submissions these days. CPV plans are encouraged because they can lead to process improvements. More specific changes would be covered by comparability protocols. With regard to criticality-assessment submissions, the panel emphasized explaining clearly how a process parameter is identified as critical and how it relates to patient risk. A deficiency often noted in submissions is that small-scale data are not linked to manufacturing-scale data.
| CMC Strategy Forum Global Steering Committee (as of July 2016) |
| Siddharth Advant (GlaxoSmithKline, previously with Celgene Corporation), Daniela Cerqueria (ANVISA, Brazilian National Health Surveillance Agency), John Dougherty (retired, previously with Eli Lilly and Company), Yasuhiro Kishioka (PMDA, Pharmaceutical and Medical Devices Agency), Junichi Koga (Daiichi Sankyo Co., Ltd.), Steven Kozlowski (CDER, FDA), Ingrid Markovic (CBER, FDA), Rohin Mhatre (Biogen), Anthony Mire-Sluis (AstraZeneca), Wassim Nashabeh (F. Hoffmann-La Roche Ltd.), Ilona Reischl (AGES, Austrian Medicines and Medical Devices Agency), Anthony Ridgway (Retired, formerly with Health Canada), Nadine Ritter (Global Biotech Experts, LLC), Thomas Schreitmüller (retired, formerly with F. Hoffmann-La Roche Ltd.), Mark Schenerman (Novavax, Inc., formerly with MedImmune, a member of the AstraZeneca Group), and Karin Sewerin (BioTech Development AB) |
Session Four: Legacy Products and Lifecycle Management
The afternoon session focused on applying lifecycle management strategies to commercial products and keeping legacy products in compliance with current regulatory expectations.
Continuous Process Verification: Thomas Damratoski of Bristol Myers Squibb (BMS) described his company’s CPV practices. It has a three-level review process for commercial products: Level 1 is each manufacturing site’s monthly review of within-batch data; Level 2 is a quarterly review across products (e.g., product yields, batch failures, percentage of batches with a process capability index (Cpk) value >1.3, and so on); Level 3 is a crossfunctional, end-to-end product review (across products), including deeper consideration of stability and product-quality statistical data in a one-slide format.
At the beginning of stage 3 (Figure 1), monitoring is risk based. BMS reviews performance plots and locks control limits after ~30 batches, applying statistical alert rules (e.g., Nelson or Western Electric rules). Cpk, time series, and histograms apply during CPV. The company uses heat maps to prioritize work and provide statistical alert event reports, which then feed data to a portfolio scorecard. Damratoski emphasized setting expectations in a quality agreement with DP contract manufacturing organizations (CMOs) to provide data and conduct statistical analyses using mutually agreed-upon approaches.
Examples of monitored DP parameters include pH, concentration, high– and low–molecular-weight (HMW, LMW) species, particulates, moisture content, impurities, and potency; glide force, stopper position, and head space are added for prefilled syringes. BMS trends DP data with corresponding DS data to distinguish sources of variation.
Useful in-process assays monitor pH, protein concentration, fill weight, and 100% visual inspection results. Bioburden and endotoxin are evaluated as binomial attributes. For fill-weight data, within-batch box plots are ideal and allow the company to check for outliers and compare manufacturing sites. Another important activity is comparing defects, major defects, and particulate defects across manufacturing sites. Damratoski warned against reporting pH data to only one decimal point when it is not distributed normally. He mentioned use of “assay variation ratio” (AVR), the ratio of reference-standard variance to batch variance in assay parameters, as a new tool for tracking assay performance. BMS considers results with Cpk <1.33 and AVR >0.45 to be high risk.
Bringing Legacy Combination Products into Compliance: Tracy TreDenick of BioTechLogic Consulting presented challenges in bringing legacy combination products into compliance with current good manufacturing practice (CGMP) and guidance for combination products (10). She began with a brief history of medical devices and combination products. Risks are assessed for the design, manufacturing, and intended uses of such devices in combination with DPs. Continuous design verification (which has concepts similar to CPV) is applied for demonstrating that a device remains in a state of control. A design history file (DHF) is compiled to document that a “design was developed in accordance with the approved design plan and the requirements of this part.”
TreDenick described the main initiatives for bringing legacy combination products into compliance. Beginning with quality systems gap analysis, a team is assigned to update systems, compile design development and control documents, define user-needs requirements, and outline remediation plans based on risk assessment. To determine use-error risk, the team searches databases for like devices on the market for complaints or recalls, the results of which could lead to redesigning a device or require documenting that its human-factor aspects are well controlled. Ultimately, a road map is built for design verification/validation using a design verification traceability matrix (DVTM) to demonstrate that design outputs meet both design inputs and user requirements. These efforts culminate in closing of the DHF. TreDenick concluded with a benchmarking to show where design control elements are placed in BLA submission sections.
Overview of Inspection Issues with Legacy Products: Two copresenters rounded out the session: Barbara Breithaupt, a CDER consumer safety officer, and Simon Pitts, an inspector in CBER’s Team Biologics office. Breithaupt presented inspection issues with legacy products, outlining detailed cases related to lacking technical knowledge and training in quality units. Pitts presented examples of FDA Form 483s relating to companies that did not investigate discrepancies, develop procedures to prevent microbial contamination with recurring out-of-trend observations, adequately document their environmental monitoring or disinfectant effectiveness studies, or meet other requirements of aseptic processing.
| CMC Strategy Forum North America Scientific Organizing Committee (as of July 2016) |
| Siddharth Advant (GlaxoSmithKline, previously with Celgene Corporation), Yves Aubin (Health Canada), John Bishop (Retired, previously with CBER, FDA), Barry Cherney (Amgen Inc.), JR Dobbins (Eli Lilly and Company), Julia Edwards (Genentech, a Member of the Roche Group, formerly with Biogen), Sarah Kennett (Genentech, a Member of the Roche Group, formerly with CDER, FDA), Joseph Kutza (AstraZeneca, formerly with MedImmune, a member of the AstraZeneca Group), Kimberly May (Immunomedics, Inc., formerly with Merck and Co., Inc.), Anthony Mire-Sluis (AstraZeneca), Stefanie Pluschkell (Pfizer, Inc.), Nadine Ritter (Global Biotech Experts, LLC), Dieter Schmalzing (Genentech, a Member of the Roche Group), Timothy Schofield (CMC Sciences, LLC, formerly with GlaxoSmithKline), Zahra Shahrokh (STC Biologics/ZDev Consulting), Jeffrey Staecker (BioPhia Consulting, Inc.), Andrew Weiskopf (Sana Biotechnology, formerly with Biogen), and Marcel Zocher (Bristol-Myers Squibb Company) |
Panel Discussion: Legacy Products, Lifecycle Management
For the final panel discussion, speakers and cochairs of the previous session were joined by Heather Eurenius of Merck.
Strategies for Bringing Legacy Products into Compliance: A major recurring compliance issue that inspectors are noticing is a lack of quality unit surveillance over manufacturing activities — with resulting potential for lack of compliance to go completely undetected. That may be due to an inadequate product/process understanding in the quality units or quality personnel’s inability to see what operators are doing.
FDA inspectors in attendance gave a clear message that they are not concerned about the number of deviations occurring, but rather the lack of them. Form 483 examples often relate to operators’ or management’s misunderstanding standard operating procedures (SOPs) or revisions thereof. Legacy-product sponsors should prioritize the quality of deviation investigations — identification of true root causes, extending investigations to other batches and other products, and so on — as well as the means of addressing them. Inspectors from both Europe and the United States said that they often bring up newer requirements during inspections and come to agreement with sponsors about the way that they will handle those changes (e.g., through annual reports or supplemental filings).
One important point brought up during these discussions related to documenting CPV trend data and knowledge gained about the sources of process variation in a deviation report. That can be used to justify decisions on investigation of a rule violation.
Strategies for Introducing Changes During CPV: Drivers for change include discovering a new critical process parameter (CPP) or introducing a new assay technology, among several others. Biosimilar developers have driven improvements in control strategies for legacy products — e.g., in process yield, cost of goods (CoG), and dosage forms. Depending on the scope and impact of a change, it could be submitted to the FDA as an annual report or as Changes Being Effected (CBE0, CBE30) report or as Post-Approval Supplement (PAS), as described in 21 CFR Part 314.70 and FDA guidance (11).
For example, a retrospective leachables and extractables study of a legacy product can be filed as an annual report. A new BLA describing a formulation change to a high-concentration DP was the driver for a Shire product that had been approved in 60 countries for 10 years. That change triggered reevaluation of quality target product profiles (QTPPs), CPPs, and control strategies. In one example of a DP manufacturing site change to a more modern facility with less process variability, a risk-based approach was used to set a tighter process control for the new site while continuing to use the same product specifications at both sites.
Specification Changes for Legacy Products: Regulatory agencies ask sponsors to adjust their specifications periodically after a product is licensed. Companies often commit to revising specifications after 25–30 lots or two years postlicensure.
Specifications often are tightened or may be kept constant. One audience member mentioned a company that had to make a shift in an in-process specification. Another represented a company that has tended to tighten input specifications for materials for better control of a process rather than change DP specifications that are tied to clinical experience, as another commenter reported. For orphan drugs and breakthrough therapies that make limited numbers of batches, specifications might be changed after review of stability data from four or five batches. Several members of the industry agreed that ample time should be given after data are locked to conduct internal assessments of process and analytical performance against clinical experience and align teams on changes to propose.
Regulators also would like to see methods updated for legacy products as new technologies are developed. This recommendation often is made during inspections. Dieter Schmalzing of Genentech raised a concern that the regulatory environment does not make such changes readily feasible. For example, it can take four years to get a release assay approved for a product that is filed in 40–50 countries across all regions; in the meantime, a company has to conduct dual testing, which is complex to execute and costly.
Compendial methods might be changed if new methods are validated as improvements. An example is subvisible particle analysis using small volumes, which was validated and implemented by some companies before USP <787> was established. In Europe, regulators expect method equivalence to be demonstrated.
Barry Cherney of Amgen mentioned that his company optimizes “right-sized testing” and has been able to reduce testing for legacy products by 40–50% based on redundancy of tests and a large body of data on them. Examples include pH, protein concentration, osmolality, sodium-dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE), and reversed-phase high-performance liquid chromatography (RP-HPLC). In one case, Amgen was able to drop DP potency testing in Europe, but US authorities have been reluctant to agree to that.
Managing global specifications thus is a major challenge. Genentech (Roche) releases products based on the tightest specifications for all markets. At Amgen, some products distributed only to certain regions can be managed with different specifications. The company also is moving toward real-time release, which would make repeated testing unnecessary for other countries.
Combination Products: Finally, discussions emphasized the regulatory expectations to validate DP and delivery devices together rather than relying on history files for empty devices to justify using them for different products. Tracy TreDenick suggested that user requirements for combination products contain a QTPP-like description of device performance in the presence of the drug. Prospective studies on delivery-device design validation should include qualification of device assembly.
With regard to inspections of combination products, US inspectors use the FDA’s Compliance Program Guidance Manual (CPGM) to tailor inspections to company operations (12). A device expert from the FDA Center for Devices and Radiological Health (CDRH) sometimes will be present. Regulators say that product reviewers generally are gaining more knowledge about devices such as prefilled syringes.
References
1 Shahrokh Z, et al. Emerging Strategies for Drug Product Comparability and Process Validation, Part 1: Analytical Tools and Drug Product Comparability. BioProcess Int. 19(3) 2021: 16–23; https://lne.e92.mwp.accessdomain.com/analytical/qa-qc/emerging-strategies-for-drug-product-comparability-and-process-validation-part-1-analytical-tools-and-drug-product-comparability.
2 CBER/CDER/CVM. Process Validation: General Principles and Practices — Guidance for Industry. US Food and Drug Administration: Rockville, MD, January 2011; https://www.fda.gov/media/71021/download.
3 TR54. Implementation of Quality Risk Management for Pharmaceutical and Biotechnology Manufacturing Operations. Parenteral Drug Association: Bethesda, MD, 2012.
4 Long M. Quality Risk Management. FDA/PDA Joint Regulatory Conference: 2014.
5 21 CFR 211.113: Control of Microbiological Contamination. US Fed. Reg. 43, 29 September 1978: 45077; amended US Fed. Reg. 73, 8 September 2008: 51932.
6 CDER/CVM. Submission Documentation for Sterilization Process Validation in Applications for Human and Veterinary Drug Products: Guidance for Industry. US Food and Drug Administration: Rockville, MD, November 1994; https://www.fda.gov/media/71442/download.
7 CBER/CDER/ORA. Sterile Drug Products Produced By Aseptic Processing — Current Good Manufacturing Practice: Guidance for Industry. US Food and Drug Administration: Rockville, MD, October 2004; https://www.fda.gov/media/71026/download.
8 CBER/CDER. Draft Guidance for Established Conditions: Reportable CMC Changes for Approved Drug and Biologics Products. US Food and Drug Administration: Rockville, MD, May, 2015; https://www.fda.gov/media/92242/download.
9 TR26. Sterilizing Filtration of Liquids. Parenteral Drug Association: Bethesda, MD, 1998.
10 CBER/CDER/CDRH/ORA. Current Good Manufacturing Practice Requirements for Combination Products: Guidance for Industry and FDA Staff. US Food and Drug Administration: Rockville, MD, January 2017; https://www.fda.gov/media/90425/download.
11 CDER. Guidance for Industry: CMC Postapproval Manufacturing Changes to Be Documented in Annual Reports. US Food and Drug Administration: Rockville, MD, March 2014; https://www.fda.gov/media/79182/download.
12 Compliance Program Guidance Manual (CPGM). US Food and Drug Administration: Rockville, MD, 18 March 2015; https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/compliance-manuals/compliance-program-guidance-manual-cpgm.
Disclaimer
The content of this manuscript reflects discussions that occurred during the CMC Strategy Forum. This document does not represent officially sanctioned FDA policy or opinions and should not be used in lieu of published FDA guidance documents, points-to-consider documents, or direct discussions with the agency.
Corresponding author Zahra Shahrokh is chief development officer for STC Biologics, 330 Nevada Street, Newton, MA 02460. Andrew Weiskopf is vice president of CMC Regulatory Affairs at Sana Biotechnology. Yves Aubin is a research scientist at Health Canada. David Allen is a research fellow for Eli Lilly. Natalya Ananyeva is a team lead at the US Food and Drug Administration’s Center for Biologics Evaluation and Research (CBER). Julia O’Neill is a distinguished fellow at Moderna Therapeutics. Nadine Ritter is president and analytical advisor at Global Biotech Experts, LLC. At the time of this conference, Siddharth Advant was executive director of biologic manufacturing at Celgene. Howard Anderson (posthumous) was team lead in the Office of Biotechnology Products at the US Food and Drug Administration’s Center for Drug Evaluation (CDER).