
JUPITER IMAGES (WWW.THINKSTOCKPHOTOS.COM)
State-of-the-art analytics guide process development by providing companies with thorough understanding, effective removal, suitable control, and comparability assessment after process changes of host cell proteins (HCPs) in recombinant biotechnology products. An array of analytical techniques and approaches can be used to establish control strategies for host cell proteins. Techniques used for HCP characterization and comparability include two-dimensional (2D) gel electrophoresis with a range of stains, 2D immunoblotting, 2D high-performance liquid chromatography (HPLC), 2D difference gel electrophoresis (DIGE), and increasingly mass spectrometry (MS) in different formats. For quality control testing of HCPs, immunoassays remain the most commonly used techniques.
The inherent complexity in analyzing a mixture made up of hundreds of HCPs with significantly different molecular characteristics at varying levels of abundance is a significant challenge to any method’s capability to detect, identify, and quantify. For example, the different affinities and avidities of antibodies raised against HCPs limit the capability of immunoassays to quantify all HCPs with equal sensitivity and accuracy. In addition, the absence of antibodies that can detect all expressed species from a host proteome can allow some HCPs to evade detection.
To bring industry, academics, and regulators together for generating integrated perspectives on the science, product quality/safety, and regulatory aspects of HCPs and their control, CASSS in January 2015 held a one-day CMC Strategy Forum in Washington, DC. Presenters’ case studies exemplified best practices and emerging analytical technologies for optimal HCP detection, including methods that maximize HCP coverage; compared the utility of commercial (generic) assays with
process-specific and platform immunoassay methods; and described MS applications to aid in HCP detection that evade immunoassays. Additionally, discussions covered current European, US, and Canadian regulatory expectations and international pharmacopeial efforts, with industry and regulatory agency representatives sharing their experiences related to product-quality risks and potential impacts on clinical outcomes. Presentation slides from this forum can be found on the CASSS website (see the “Forum Talks” box). Here we summarize the key discussion points that critically evaluated both existing and emerging technologies for characterization, clearance, comparability, and quality control of HCPs in biotechnologyderived products.
| CMC Forum Series The CMC Strategy Forum series provides a venue for biotechnology and biological product discussion. These meetings focus on relevant chemistry, manufacturing, and controls (CMC) issues throughout the lifecycle of such products and thereby foster collaborative technical and regulatory interaction. The forum strives to share information with regulatory agencies to assist them in merging good scientific and regulatory practices. Outcomes of the Forum meetings are published in this peer-reviewed journal to help assure that biopharmaceutical products manufactured in a regulated environment will continue to be safe and efficacious. The CMC Strategy Forum is organized by CASSS, an International Separation Science Society (formerly the California Separation Science Society), and is supported by the US Food and Drug Administration (FDA). |
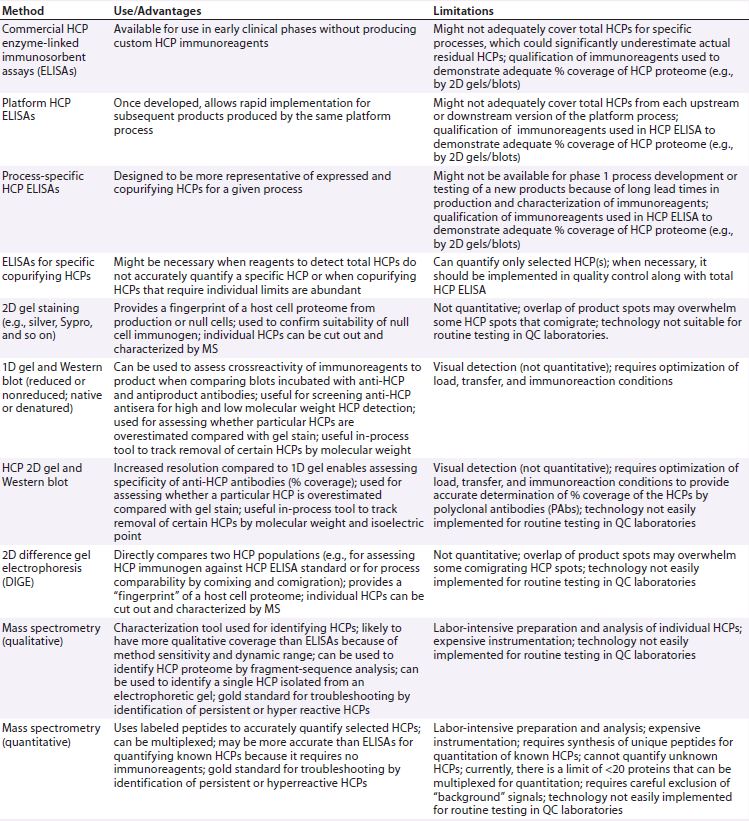
Table 1: Analytical tools for host cell protein (HCP) analysis
Analytical Tools for HCP Analysis
Table 1 lists the types of assays currently used for analysis and characterization of HCPs. Currently enzyme-linked immunosorbent assays (ELISAs) remain the “gold standard” for routine quality control (QC) release testing. Because a multianalyte ELISA generates a summed HCP response for samples, users cannot distinguish which HCPs are present in their samples and at what level each may be present. But this type of assay serves as a relatively simple method for QC operations that can be validated for current good manufacturing practice (CGMP) compliance. It also yields a single, reportable HCP value for setting specification limits.
Orthogonal methods such as 2D gels, 2D-HPLC, or MS can be used to characterize HCPs. For instance, MS can identify specific HCPs or assess similarity of HCP profiles following process changes. But these techniques are currently too complex operationally for routine QC testing. As MS-based assays evolve (e.g., to provide simplified or automated analysis), it may be possible to validate them in the future for QC release testing. In principle, any assay format could be used to control HCPs if it is demonstrated to measure them accurately and sensitively and if it can be validated for reliable performance. But it is recommended to discuss a new HCP approach for QC release with your regulatory agency before implementing.
Multianalyte ELISAs face two key challenges when used for HCP analysis:
- The immunogen starting material used to generate anti-HCP antibodies and for use as the reference standard in an HCP ELISA may not contain all the HCPs (analytes) that could be present in a drug substance.
- The wide range of abundance, affinities, and avidities of a mixed polyclonal antibody (PAb) population generated against an HCP immunogen preparation could lead to over- or underestimation of any given HCP in a drug substance.
A critical aspect of ELISA assays for quantitation of HCP levels is the overall specificity or coverage of PAb reagents used in HCP ELISAs to detect the broad range of potential HCPs in a given drug substance. Development of such an ELISA for a biopharmaceutical product requires broadly specific HCP immunoreagents derived from its expression system and manufacturing process. Therefore, it is critical to establish which HCPs from the host proteome generated by the production cells can be detected (or missed) by a PAb assay reagent.
Pharmacopoeial Efforts
A draft USP chapter on HCP measurement was finalized in the Second Supplement to USP38-NF33 in summer 2016 (1). A European general chapter on HCP assays was adopted in March 2016 and will be published in Supplement 9.1 of the European Pharmacopoeia this fall — it will be effective in April 2017, and the draft version can be found online now (2). These documents provide guidance on selection, development, and validation of HCP assays and describe specific considerations for process-specific, platform, and generic assays. Presentations, case studies, and discussions at this CMC Strategy Forum paralleled the principles captured in those general chapters.
Recommended Preparation Methods for Antigen and Antibody Reagents with High HCP Coverage: Typically a cell culture fluid or cell lysate of a null host cell line (without the transfected product plasmid) is grown under manufacturing process conditions representative of the drug substance, then used as the immunogen to produce PAbs reacting with a broad range of potential HCPs. The most common species used for production of anti-HCP antisera are goats and rabbits, but other species are acceptable. Use of antiserum from a single animal or multiple animals, or pooling of antisera generated against separate immunogens, has been successful in producing high-coverage ELISA reagents. Pooling antisera from different species might increase the likelihood of obtaining a diverse set of anti-HCP antibodies.
Using Western blot analysis, each antiserum can be assessed for titer level against the total HCP proteome; then the highest-titer, broadest-coverage antisera are pooled. That provides larger quantities of the mixed PAb reagent. Having a supply of HCP immunoreagents that lasts through the lifecycle of a product (e.g., 10–20 years) is desirable. Companies that do so won’t have to revalidate reagents with subsequent refilings of their HCP ELISAs, which carries a risk of not being able to demonstrate similar results as those obtained by earlier immunoreagents. To generate a large supply of HCP immunoreagents, sera from either five to eight large animals (e.g., goat) or 20–30 smaller animals (e.g., rabbits) typically are pooled.
There are different approaches to the preparation of purified PAbs from antisera. The two major strategies are affinity purification of total immunoglobulin G (IgG) over affinity columns with immobilized protein G or A; or affinity purification of only anti-HCP antibodies over HCP-immobilized columns. The first approach yields an immunoreagent containing nonspecific antibodies in addition to specific anti-HCP PAbs, whereas the second approach captures only those anti-HCP PAbs that recognize HCP antigens bound to the affinity column.
Regardless of the immunization or purification strategy chosen, both regulators and industry consider it critical to characterize the resulting anti-HCP reagent’s degree of immunospecificity. Key characterization elements include
- assessing the reagent’s lack of cross-reactivity with the protein product itself
- assessing its ability to detect low-abundance and/or low–molecular-weight HCP species
- assessing the detection of HCP subpopulations that are enriched in downstream steps
- demonstrating the percentage of the total host cell proteome that the PAbs can detect.
Western blots visually demonstrate that degree of immunospecificity. 2D sodium-dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) separation of complex protein mixtures according to their isoelectric point and molecular weight, followed by Western blotting with the anti-HCP PAbs, can provide insight into the PAbs’ immune coverage with respect to HCPs in the immunogen used for generating them. To investigate product cross-reactivity, scientists compare 1D or 2D Western blot developed with anti-HCP PAbs with a second blot developed with antiproduct antibodies. This approach also can be used to differentiate HCP bands from “product-related” bands observed on a gel.
Currently the “gold standard” method to show the degree of immunorecognition of anti-HCP PAbs for the total HCP population is 2D gel staining with Western blotting, which compares the number of gel spots detected by a sensitive staining method with those that are also recognized by anti-HCP PAbs in a Western blot analysis. It is generally recognized that such an approach can miss detecting some HCPs: Gels typically reduce and denature HCPs, which can result in a loss of epitopes that are solely conformational (although it is believed that conformational-only epitopes could be a small subset of total HCP epitopes). And for each HCP and PAb reagent set, gel/blot conditions require experimental optimization of the load and stain development time to enable accurate results. A low sample load can underestimate the total HCPs present in the host cell proteome; a high sample load can lead to artificial nonspecific reactivity of anti-HCP antibodies to the product itself.
To supplement 2D gel/blot specificity data, developers can characterize HCP ELISA reagents with orthogonal methods, including 2D HPLC, antibody affinity extraction, and MS. Because such analytical characterization data sets demonstrate the immunospecificity of the HCP ELISA to detect residual HCPs (which is the key intended use of that method), they should be presented to demonstrate specificity in HCP ELISA method-validation package in regulatory submissions.
Forum Discussion
How to Overcome Dilutional Nonlinearity in Immunoassays with Broad HCP Coverage: Anomalous binding effects present one of the most technically challenging aspects of using a highly diverse set of PAbs in a single ELISA well. A common problem is an excess of one or more HCP antigens in test samples that overwhelm available anti-HCP antibodies in the capture or detection reagents. This is seen as a “hook effect” in the results, when higher dilutions of test material yield apparently higher HCP levels. In some cases, the hook manifests as a plateau level, followed by linearly decreasing HCPs with increasing dilution of the test material. But in other cases, a plateau level is never reached.
HCP ELISA developers need to evaluate those phenomena experimentally for each assay and test sample type (e.g., process intermediates), then validate the minimal valid dilution (1, 2) at which the HCP level (after correction for sample dilution) is independent of sample dilution (demonstrating dilutional linearity). However, the risk of using sample dilution to overcome the hook effect is that it also dilutes the HCPs so that their levels could be underestimated for a given product batch.
When an HCP copurifies with a drug substance, it may be necessary for that HCP to be characterized for its identity. If it is determined to be a risk, then it should be subjected to an appropriate control strategy. In other circumstances, some HCPs might be present at such high abundance that they effectively prevent detection of other, lower-abundance HCPs. This effect has occurred with the highly abundant milk proteins (e.g., casein or lactalbumin) in products generated by transgenic mammals. Such proteins swamp out the signal from other HCPs that are at much lower levels. In such cases, a separate HCP assay (such as a protein-specific ELISA) often is developed to ensure that those individual HCPs will be controlled within their clinically qualified limits.
How to Replace and Bridge the HCP ELISA Critical Reagents: It is a best practice to prepare a sufficient quantity of HCP immunogen and anti-HCP PAbs that will last through a product’s planned lifetime, thus eliminating the need to replace such complex protein mixtures. If that is not possible (e.g., for legacy products), then it will be necessary to bridge a new immunoreagent set with sufficient analytical characterization of the immunogen and antibodies to show similar capability to that of the original reagents for detecting and quantifying HCPs. Changes in process-specific immunoreagents should be assessed for sensitivity and specificity to assure continuity of data. Thus, preparation and characterization of HCP ELISA reagents (immunogen, antisera, and PAbs) should be thoroughly documented with sufficient technical details to allow them to be recreated when supplies become depleted. Because an HCP ELISA result is a sum of all immunoreactions and thus dependent on both the level and affinity of different HCP/antibody pairs, it alone cannot compare the specificity of mixed populations in original and new immunoreagents. Therefore, bridging of new HCP immunoreagents requires more than just testing product batches in an HCP ELISA assay.
A bridging study to assess the suitability of the new HCP reagents should use methods that compare the pattern and abundance of proteins in the HCP ELISA standard or the immune-specificity of the anti-HCP PAbs (or both, if both reagents are replaced). At a minimum, a bridging study would include a repeat of 2D gel staining with Western blotting to compare old and new immunoreagents. 2D-HPLC may supplement the blot data. Changes made only to the HCP mixture may be assessed with comparative 2D-DIGE and/or MS pattern comparison of old and new standards. In addition, the HCP coverage of in-process samples and drug substance by the old and new reagents would be assessed.
Additional bridging data should then come from the HCP ELISA by demonstrating dose response parallelism of the old versus the new HCP standard/antibody sets. Finally, representative samples of process intermediates and of drug substance lots should be tested by the two reagent sets to check if the ELISA values are comparable.
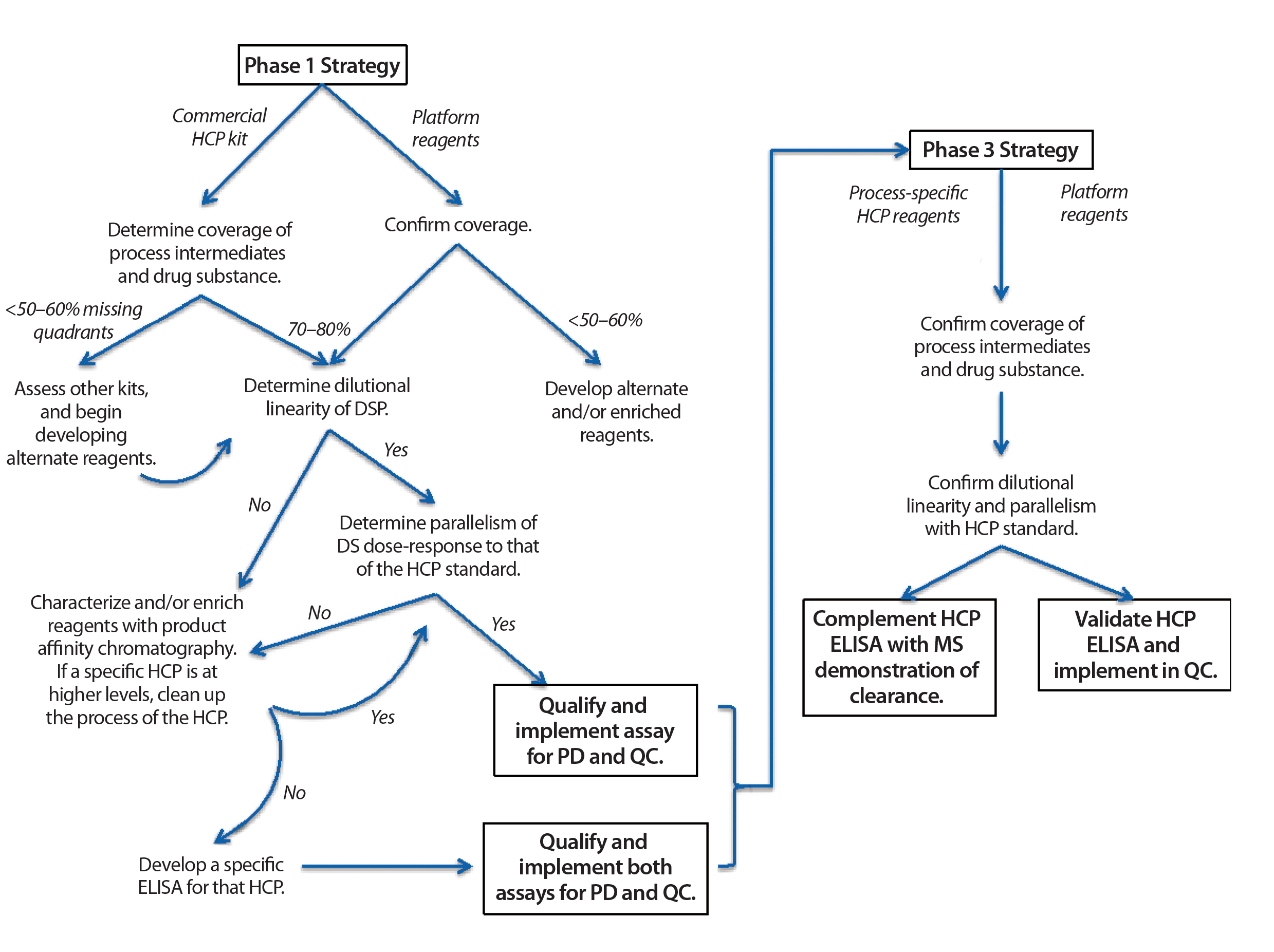
Figure 1: Guide to phase-appropriate host-cell protein assay development
How to Determine Suitability of HCP Assays at Different Product Lifecycle Stages: Figure 1 shows the typical evolution of HCP ELISA reagent development over a product’s lifecycle. Because it can take a year to generate a process-specific set of HCP reagents, often a commercially available ELISA assay reagent set is used (if such reagents are available for a given product’s host cell) in early development. Such assays provide preliminary data on process capability and product quality. Even when developers use commercial HCP ELISAs in early phases, we recommend that their reagents be evaluated by 2D gels/blots to assess for specificity toward and coverage of the proteome of the host cell. Additionally, users should check the cross-reactivity of commercial antibody reagents to a protein product itself by Western blot. Resulting data will provide critical information on what is and is not being detected by the commercial ELISA, which is important for guiding process development studies later on.
Difficulties with commercial ELISAs include an inability to detect the same signals in upstream and downstream materials and kits from different manufacturers detecting different HCP populations in the same product. In one example, three generic HCP ELISA kits and one process-specific HCP ELISA were used to compare five batches of an originator product relative to six batches of a biosimilar product. Large differences were seen in the types and levels of HCPs detected, depending on which assay reagent was used. Hence, it might be prudent to evaluate commercial kits from different vendors in early clinical development and identify one with maximal coverage of the HCPs for a given product. Observing poor coverage with commercial ELISA reagents might trigger an earlier start on developing process-specific ELISA reagents. It also might necessitate use of orthogonal methods that do not require immunoreagents (e.g., 2D DIGE) to provide supplemental data on what is not being detected by that HCP ELISA.
US regulators generally accept the use of commercially available HCP ELISA assays during preclinical development. They also may be used during early stages of clinical trials if assay coverage is demonstrated. However, switching to a process-specific ELISA as early as possible is the best means for derisking process development and process validation/performance qualification. Representatives of the US Food and Drug Administration (FDA), Health Canada, and European regulators all indicated at this forum that by the time of biologics license application (BLA) or market authorization application (MAA) filing, a suitable processpecific HCP ELISA should have been developed and implemented. Regulators typically expect companies to do that before entering phase 3 clinical trials. When HCP ELISA dilutional linearity is poor, studies should be conducted to identify whether one or more specific proteins are contributing to that behavior. Such HCPs should be identified by MS characterization or other appropriate methods, and a strategy then can be proposed to either remove those species from the process or monitor them with specific ELISAs.
To set meaningful HCP specifications after making changes to HCP ELISA reagents, it is critical for a company to archive samples of all clinical drug-substance batches that were tested with an earlier set of reagents and retest those with the new process-specific assay. The new reagents could yield residual HCP levels that are higher than those from commercial reagents because of greater HCP detection. But those values will represent a more accurate measure of clinically qualified residual HCP levels and thus are suitable for establishing specification limits.
Questions and Answers
What HCP comparability studies should be performed to support process changes? Even process-specific HCP ELISAs may not detect all HCPs from a given host cell. So to support comparability studies following process changes, the best practice is to supplement HCP ELISA analysis of batches made by the original and new processes with analysis by broad-based, reagent-independent characterization methods such as MS, 2D gels, and/or 2D-DIGE. Those methods are used to assess whether new HCPs have been introduced with the new process and whether the level of any individual HCP has significantly increased following process change.
What are the regulators’ current thoughts on establishing a commonly acceptable HCP limit in biosimilar products? No common HCP limits can be officially set for all products because limits are based on factors such as product indication (dose, route of administration, and patient population), clinical purpose (chronic, replacement, or acute therapies), and variances in HCP test methods (specificity and sensitivity) and the types of HCPs present. Moreover, not all HCPs are equally problematic. In some products, a single specific HCP has caused adverse events, whereas many legacy protein products containing rather high HCP levels did not show adverse clinical effects.
A broad range (from low nanogram to low microgram) measured by different units (ppm, ng/mg, ng/unit) has been reported for HCP levels in recombinant products. For commercial monoclonal antibody therapeutics (MAbs), limits of 1 ng/mg (1 ppm) can be reached after process optimization. It is not unusual for developers of recombinant therapeutics to propose a phase 1 HCP specification limit of 100 ppm, typically using a commercial HCP ELISA.
For biosimilar products, developers cannot simply compare HCP levels to marketed originator products because different sets of immunoreagents are used to establish those products’ HCP specifications. Thus, an HCP specification is set for each biopharmaceutical product based on clinically qualified ranges supported by demonstrated process consistency. Although a quantitative HCP level cannot be used to demonstrate comparability of biosimilars and originator products, some regulatory agencies are currently considering the use of MS data to compare “HCP profiles” in such cases. A recent proteomics approach analyzing protein-A–purified commercial-grade MAbs to remove a product of interest quantified 700 HCPs at 1–10 ppm levels. Multivariate analysis enables comparison of HCP profiles for products from various sources. That may be one approach for assessing HCP profile comparability of biosimilars and originator products.
In some cases, an appropriately specific HCP ELISA is used, HCP spiking studies using small-scale models show adequate capability of the process to remove HCPs, and testing of sufficient commercial drug-substance lots (e.g., 10–30 lots) continues to yield HCP levels at or below the established limits. Regulators then might allow HCP testing to be removed from a commercial product-release specification, although it would remain necessary for use in characterization and comparability studies conducted after product approval.
What common HCPs should be of particular concern for a given class of products or cell line? Forum presenters showed several examples of copurifying HCPs that have been identified to remain tightly associated with biotechnology-derived proteins. Analysis of HCPs that bind to protein A resin (only in the presence of MAbs) has indicated different subpopulations of HCPs in Chinese hamster ovary (CHO) cell lines that bind to different MAbs through electrostatic or hydrophobic interaction. The presence of such proteins is sometimes manifested by dilutional nonlinearity or nonparallelism of sample dilutions compared with an analytical HCP standard.
One such CHO protein is phospholipase B-like 2 (PLBL2), which has been observed at different levels in MAbs produced using platform processes. Levels >10 ng/mL caused nonlinear dilutions in a platform HCP ELISA assay. Recently, degradation of polysorbate surfactant used in protein formulations was found to result from the presence of trace phospholipase levels in MAbs produced by CHO cells and enzymes from a human cell line. Thus we recommend checking for the adequate clearance of phospholipases when developing recombinant proteins from such sources.
Other examples of copurified HCPs include Escherichia coli host proteins OppA and DppA, which require hydrophobic-interaction chromatography to separate them from apolipoprotein AI products and glutathione synthase that was not detected by a platform HCP ELISA, but was detected by SDS-PAGE Western blotting. Copurifying HCPs causing dilutional nonlinearity have been observed in multiple replacement therapies produced by the same human cell line at the same manufacturer, yet neither one of the identified HCPs was the same among those different products. These observations point to a need for thorough evaluation of each protein product’s HCP content even with platform products and processes.
An unusual case presented at the forum was that of a product expressed by a customized proprietary CHO cell line that elicited anti-HCP antibodies in patients during clinical trials. The early phase process was developed using SDS-PAGE, reverse-phase (RP) HPLC, and Western blot to determine HCP levels along with a commercial HCP ELISA kit. Patient anti-HCP antibodies were used for affinity chromatographic purification of the immunogenic HCPs, which were then identified by LC/MS/MS analysis. A combination of multiple-reaction monitoring MS (LC-MRM) and Western blot methods guided development of a revised purification process. The company developed a process-specific set of ELISA reagents from a null cell line, using both the LC-MRM and process-specific ELISA methods were demonstrating consistent removal of the immunogenic HCPs in a revised commercial process. Although the presence of anti-HCP antibodies did not correlate with clinical safety issues, the program was placed on clinical hold until resolution of this issue.
What is the regulatory perspective on risk evaluation of HCPs in biological products? Regulators evaluate clinical risk of HCPs based on the following factors:
- Dose and frequency (the higher or more frequent the dose being of greater concern than a single low dose of a drug because of the higher amount of total HCPs that is delivered)
- Patient population and product class (pediatrics/healthy or terminally ill patients and immune-stimulatory or immunesuppressive products; e.g., risk of a cytokine treatment in a pediatric population may be of a greater concern than an anticancer MAb in terminal cancer patients, for whom benefits outweigh risks)
- Route of administration (different risk for subcutaneous and intravenous administration, with its potential to trigger immune responses)
- Host cell source (anaphylactic reactions have been observed with yeast-derived vaccines; the potential homology of an HCP sequence to a human protein that might generate anti-HCP antibodies that could trigger autoimmunity)
- Safety signals (from nonclinical and clinical data)
- HCP levels (adjuvant effect, with high host cell protein levels in early phase manufacturing process lots causing high immunogenicity in patients)
- Activity of a specific HCP (intrinsic bioactivity; e.g., host cell proteases induce degradation of protein product and host cell lipases cause degradation of polysorbate excipients).
When reviewing a dossier, regulators look for overall control of HCPs as a critical quality attribute. Elements of control include manufacturing process consistency in efficiently removing HCPs, appropriate safety-based HCP specification limits based on clinical experience, and suitability of analytical methods used for monitoring HCPs (coverage, sensitivity, specificity, source of immunogen, and degree of characterization with orthogonal methods). An upstream process intermediate is considered a more suitable immunogen than a downstream process intermediate because it can provide broader coverage and allow for better assessment of HCPs following process changes.
What is the regulatory thinking about routinely monitoring anti-HCP antibodies during clinical development? For biologically derived products, regulatory agencies expect immunogenicity testing for antidrug antibodies (ADAs), which may include screening for anti-HCP antibodies depending on the level of product risk (see above). High-titer and persistent anti-CHO antibodies have been observed in patients dosed with a CHO-derived clotting factor product. Thus, for replacement therapies with recombinant plasma proteins (that tend to be of high concentration and frequent dosing), the Office of Blood Research and Review at the FDA’s Center for Biologics Evaluation and Research (CBER) indicated that it is currently requesting sponsors to develop clinical immunogenicity assays for screening anti-HCP antibodies during phase 1 clinical studies as part of their ADA studies. The FDA’s Center for Drug Evaluation and Research (CDER) stated that it will continue to approach anti-HCP antibody assays case by case using risk assessment based on the principles of a 2014 guidance on immunogenicity assessment (3).
Early Knowledge is Key
One regulator commented in our forum discussions, “HCP assays are currently the weakest analytical link in the total characterization and control strategy for biotech and biosimilar products.” Given the complexities touched on herein, verifying the suitability of an HCP assay in early clinical development and using orthogonal characterization
methods to better understand HCPs in the process and the product can help companies prevent delays, rework, and potential risk to patients.
Acknowledgments
As the scientific committee members of this forum, we are especially grateful to our speakers — Martin Schiestl (Sandoz), Mary Vanderlaan (Genentech, a Roche Company), Kevin Van Cott (University of Nebraska), Tony Mire-Sluis (Amgen, representing USP Expert Panel), Laurie Graham (CDER), Alexey Khrenov (CBER), and Jörg Engelbergs (Paul-Ehrlich-Institut) — and additional panel members — Tracy Burton (Health Canada), Phoebe Baldus (Pfizer), Roman Drews (LFB-USA), Ken Hoffman (Cygnus Technologies), Girija Krishnamurthy (BristolMyers Squibb), and Gwenaël Cirefice (EDQM) — for sharing their valuable experiences.
Forum Talks
| Find these CMC Strategy Forum speaker presentations online at http://casss.site-ym.com/general/custom. asp?page=CMCJ1513. |
| Bishop J, Meiklejohn B. Best Practices and Emerging Analytical Technologies to Achieve Optimal Detection of Host Cell Proteins in Process and Product. |
| Graham L, Khrenov A. Risk Assessments and Control Strategies for Host Cell Proteins: FDA Expectations and Experiences. |
| Ho K, Mire-Sluis A. EDQM and USP Guidance Documents for Host Cell Proteins: Convergence and Differences. |
| Rawat R, Sluzkey V. Regulatory Expectations for Host Cell Protein Testing and Control |
| Van Cott K. Retrospective Analysis of a Host Cell Protein Perfect Storm: Identifying Immunogenic Proteins and Fixing the Problem. |
| Vanderlaan M. Hamster Phospholipase B-Like 2 (PLBL2): A Host Cell Protein Impurity in CHO-Derived Therapeutic Monoclonal Antibodies. |
| Wolschin F, Schiestl M. Host Cell Protein Analysis By Mass Spectrometry and Its Application in Comparability Exercise. |
| North American Program Committee for These Forums |
| Siddharth Advant (Kemwell Biopharma), Yves Aubin (Health Canada), John Bishop (FDA-CBER), Barry Cherney (Amgen Inc.), JR Dobbins (Eli Lilly and Company), Julia Edwards (Biogen Idec), Sarah Kennett (FDA-CDER), Joseph Kutza (MedImmune, a member of the AstraZeneca Group), Kimberly May (Merck & Co., Inc.), Anthony Mire-Sluis (Amgen Inc.), Stefanie Pluschkell (Pfizer, Inc.), Nadine Ritter (Global Biotech Experts, LLC), Reb Russell (Bristol-Myers Squibb Company), Oscar Salas-Solano (Seattle Genetics, Inc.), Dieter Schmalzing (Genentech, a member of the Roche Group), Timothy Schofield (MedImmune, a member of the Astra Zeneca Group), Zahra Shahrokh (STC Biologics, Inc. and ZDev Consulting), Jeffrey Staecker (Genzyme Corporation, a Sanofi company), and Andrew Weiskopf (Biogen Idec) |
Disclaimer
| The content of this manuscript reflects the group discussions that occurred during the CMC Strategy Forum, including possible points of disagreement among attendees. Further, this document does not represent official FDA policy or opinions, and should not be used in lieu of published FDA guidance documents, points-to-consider guidance documents, or direct discussions with the agency. |
References
1 USP <1132> Residual Host Cell Protein Measurement in Biopharmaceuticals. Second Supplement to USP38–NF33, June 2016.
2 2.6.34: Host Cell Protein Assays. Pharmeuropa 27(2) 30 March 2015; www.e-gmp.pl/wp-content/uploads/2015/05/2.6.34.Host-cell-protein-assays-draft.pdf.
3 CBER/CDER. Guidance for Industry: Immunogenicity Assessment of Therapeutic Protein Products. US Food and Drug Administration: Rockville, MD, 2014; www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm338856.pdf.
Supplier Technical Notes
Application Note 28-9794-25 AA: Characterizing Host Cell Protein Patterns with 2-D DIGE Improves Process Understanding. GE Healthcare: Uppsala, Sweden, 2010. www.gelifesciences.com/gehcls_images/GELS/Related%20Content/Files/1314807262343/litdoc28979425_20140202232039.pdf.
Bishop E, Hoffman K. Affinity Antibody Extraction. Cygnus Technologies: Southport, NC; www.funakoshi.co.jp/download/catalog/CYG5209.pdf.
Berkelman T, Harbers A, Bandhakavi S. Tech Note Bulletin 6393, Rev A: Reliable, Streamlined 2-D Western Blot Workflow for Evaluation of Antibodies Developed for Detection of Host Cell Proteins. BioRad Laboratories, Inc.: Hercules, CA 2013; www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6393.pdf.
Cygnus Technologies Inc. White Paper: Non-Specific Binding In Western Blots. Bioprocess Online 8 July 2011; www.bioprocessonline.com/doc/non-specific-binding-in-western-blots-0001.
Further Reading
Aboulaich N, et al. A Novel Approach to Monitor Clearance of Host Cell Proteins Associated with Monoclonal Antibodies. Biotechnol. Progr. 30(5) 2014.
Catalin E, et al. Analysis of Host-Cell Proteins in Biotherapeutic Proteins By Comprehensive Online Two-Dimensional Liquid Chromatography/Mass Spectrometry. MAbs 4(1) 2012: 24–44.
Catherine EM, et al. The Dynamics of the CHO Host Cell Protein Profile During Clarification and Protein A Capture in a Platform Antibody Purification Process. Biotechnol. Bioeng. 110(1) 2013: 240–251.
Champion K, et al. Defining Your Product Profile and Maintaining Control Over It, Part 2: Challenges of Monitoring Host Cell Protein Impurities. BioProcess Int. Sep 2005: 52–57.
Dixit N, et al. Residual Host Cell Protein Promotes Polysorbate 20 Degradation in a Sulfatase Drug Product Leading to Free Fatty Acid Particles. J. Pharm. Sci. 105(5) 2016: 1657–1666.
Hall T, et al. Polysorbates 20 and 80 Degradation by Group XV Lysosomal Phospholipase A2 Isomer X1 in Monoclonal Antibody Formulations. J. Pharm. Sci. 105(5) 2016: 1633–1642.
Hunter AK, et al. Separation of Product Associating E. coli Host Cell Proteins OppA and DppA from Recombinant Apoliloprotein A-IMilano in Industrial HIC Unit Operations. Biotechnol. Progr. 25(2) 2009: 446–453.
Ingerslev J, et al. Antibodies to Heterologous Proteins in Hemophilia A Patients Receiving Recombinant Factor VIII (Recombinate). Thromb. Haemost. 87(4) 2002: 626–634.
Jin, et al. Profiling of Host Cell Proteins By Two-Dimensional Difference Gel Electrophoresis (2D-DIGE): Implications for Downstream Process Development. Biotechnol. Bioeng. 105(2) 2010: 306–316.
Levy NE, et al. Identification and Characterization of Host Cell Protein Product-Associated Impurities in Monoclonal Antibody Bioprocessing. Biotechnol. Bioeng. 111(5) 2014: 904–912.
Madsen J, et al. Toward the Complete Characterization of Host Cell Proteins in Biotherapeutics Via Affinity Depletions, LC-MS/MS, and Multivariate Analysis. MAbs 7(6) 2015: 1128–1137.
Reisinger V, et al. A Mass Spectrometry–Based Approach to Host Cell Protein Identification and Its Application in a Comparability Exercise. Anal. Biochem. 463, 2014: 1–6.
Savino E, et al. Development of an In-House, Process-Specific ELISA for Detecting HCP in a Therapeutic Antibody, Part 1. BioProcess Int. March 2011: 38–48.
Tait AS, et al. Host Cell Protein Dynamics in the Supernatant of a mAb-Producing CHO Cell Line. Biotechnol. Bioeng. 109(4) 2012: 971–982.
Vanderlaan M, et al. PLBL2: A Host-Cell Impurity in Therapeutic Monoclonal Antibodies Derived from Chinese Hamster Ovary Cell. BioProcess Int. 13(4) 2015: 18–55.
Wang X, et al. Host Cell Proteins in Biologics Development: Identification, Quantitation, and Risk Assessment. Biotechnol. Bioeng. 103, 2009: 446–458.
Wang X, et al. Improved HCP Quantitation By Minimizing Antibody Cross-Reactivity to Target Proteins. BioProcess Int. Jan 2010: 18–24.
Zhu-Shimoni J, et al. Host Cell Protein Testing By ELISA and the Use of Orthogonal Methods. Biotechnol. Bioeng. 111(12) 2014: 2367–2379.
John Bishop is project manager at CBER, FDA; john.bishop@fda.hhs.gov. Jörg Engelbergs is a scientific assessor in quality and nonclinic at Paul-Ehrlich-Institut; joerg.engelbergs@pei.de. Erika Friedl is scientific assessor in quality at Paul-Ehrlich-Institut; frier@pei.de. Kowid Ho is in technical regulatory (CMC) policy at F. Hoffmann-La Roche Ltd.; kowid.ho@roche.com. Bruce Meiklejohn is a senior research fellow in CMC regulatory biotechnology at Eli Lilly and Company; bim@lilly.com. Rashmi Rawat is a biologist at CDER, FDA; rashmi.rawat@fda.hhs.gov. Nadine Ritter is president and senior analytical advisor at Global Biotech Experts, LLC; nadine.ritter@globalbiotechexperts.com. Dieter Schmalzing is senior principal technical advisor at Genentech, a Member of the Roche Group; schmalzing.dieter@gene.com. Zahra Shahrokh is chief development officer at STC Biologics; zshahrokh7@gmail.com. Victoria Sluzky is group vice president of quality and process development at BioMarin Pharmaceutical Inc.; vsluzky@bmrn.com.