FDA approval of Teva’s fremanezumab depends on a speedy resolution of a plant run by CMO Celltrion but this is looking increasingly likely, an analyst says.
The US Food and Drug Administration (FDA) had set the Prescription Drug User Fee Act (PDUFA) action date for fremanezumab, a humanized monoclonal antibody targeting calcitonin gene–related peptide (CGRP) for mid-June.
But Teva Pharmaceutical Industries envisioned a delay after its contract manufacturer Celltrion received an FDA warning letter in January.
Celltrion’s site in Incheon, South Korea makes the active pharmaceutical ingredient (API) for the proposed migraine treatment. Teva now expects a regulatory decision to be made for its monoclonal antibody candidate by September 16.
However, this will depend on the FDA’s satisfaction with the site, and would mean Celltrion resolving the GMP issues – and undergo a successful reinspection – in less than six months from receiving the warning.
5 Month Resolution
Despite this, Evercore ISI analyst Umer Raffat wrote it “seems increasingly likely that Teva can get an approval for the CGRP in 2018” in a note to investors.
The FDA directed its criticisms to the fill & finish plant, and not the production suite from where fremanezumab API is made. But the Federal Food, Drug, and Cosmetic (FD&C) Acts – Section 501(a)(2)(B) – states there is no distinction between API and finished pharmaceuticals when defining whether a facility is in CGMP compliance.
“TEVA is likely flagging to FDA that Celltrion’s API is in good shape. With that said, I still think that fill/finish can’t continue to be a warning letter state for Teva to get an approval.”
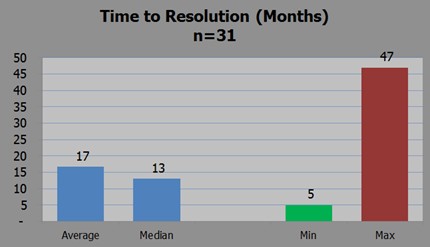
Warning letter resolution time. Image c/o Umer Raffat, Evercore ISI
However, he also analyzed 31 warning letters relating to CGMP issues on finished pharmaceuticals and concluded warning letter on fill/finish issues have been resolved in as little as 5 months, although that is the best outcome.
Furthermore, Raffat noted TEVA is using a priority review voucher, which may aid in expediting an FDA reinspection.
And a further indication of good news for Teva is the FDA’s choice to issue a PDUFA extension, rather than issuing a complete response letter (CRL).
In April, the FDA issued CRLs to versions of Rituxan (rituximab) and Herceptin (trastuzumab) due to the manufacturing deficiencies at the plant. Teva inked a North American commercialization partnership with Celltrion for the two biosimilars in 2016.