Because of the molecular complexity and relative fragility of biotherapeutics, validated intermediate hold times are critical for their commercial manufacture. Manufacturers typically conduct studies to define acceptable hold times for process intermediates to determine acceptable hold times for in-process production samples. A validation study defines maximum allowable hold times for all intermediate process stages based on product-specific data obtained during a hold study. A systematic risk assessment can determine which intermediate hold points should be validated (1).
Hold materials are typically obtained from production-scale batches and held at set temperatures and times. Time points are sampled periodically and assayed for product quality attributes (e.g., aggregates, fragments, oxidation, and acidic species) that may be affected by the hold period and for microbial control (e.g., bioburden and endotoxin) (2). Intermediate hold studies are often executed during large-scale manufacture of a biopharmaceutical after proof of concept, ideally shortly before or during performance qualification batches to ensure that process parameters are fixed and that changes implemented will not invalidate a hold study.
PRODUCT FOCUS: ALLBIOLOGICS
PROCESS FOCUS: INTERMEDIATE STAGES
WHO SHOULD READ: PrOCESS DEVELOPMENT AND MANUFACTURING
KEYWORDS: SAMPLING METHODS, SAMPLING STORAGE, CONTAINMENT SYSTEMS
LEVEL: INTERMEDIATE
One recent publication describes a hold-time study to validate an acceptable time for a low-pH hold point during production of a representative monoclonal antibody (MAb) (3). In principle, the process of validating intermediate hold points appears relatively straightforward; however, the complexity of the study, operational constraints, and the time-dependent coordination required among functional groups are often underestimated.
Selecting Scaled-Down Vessels and Storage Conditions
Although one approach is to conduct intermediate hold studies at scale within a production environment, it can be costly, time consuming, and logistically challenging because material may need to be held both at 2–8 °C and ambient temperature for different time points. Because of operational constraints and affect on a production standard work schedule, usually hold material is transferred to a QC or TS laboratory and aliquotted, and samples are held at small scale.
During a study’s planning phase, appropriate small-scale vessels should be selected. Low-volume bioprocess bags are commercially available with the same materials of construction and contact-surface layers. Small-scale 316L-grade stainless steel vessels also can be sourced (Photo 1) and filled to represent an equivalent ratio of volume to head space as that observed at production scale.
Head-space ratio is important because it may affect product quality attributes. Increased head space combined with agitation in a harvest hold vessel can increase product oxidation during a hold period. Production samples held within stainless steel vessels might be agitated during a hold study, although the method of agitation, stirring rate, and gas transfer can be difficult to accurately scale down. Hence a nominal agitation rate can be selected to reflect an approximate rate of mixing in a vessel during its hold period. In addition, worst-case hold temperatures can be considered and samples held within temperature-mapped and calibrated incubators or cold rooms. Consecutive hold samples can be taken from a single bulk production sample held under appropriate conditions. Alternatively, they can be aliquotted and stored within a number of scaled-down stainless steel vessels or bioprocess bags — with each vessel or bag representing a defined hold point.
Sampling and Materials Transfer
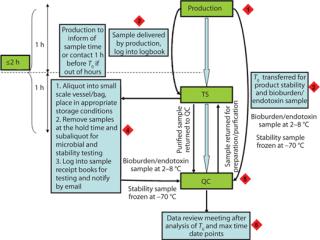
Intermediate hold samples are typically held within a laboratory environment. Bulk samples should be transferred from production at the end of discrete unit operations that are considered “hold points.” Sample tracking with visibility boards and daily sample schedules is one effective approach to ensure that samples are taken and transferred from production at appropriate times (Figure 1). Tracking is important because the times when samples are taken and made available for collection within production may be approximate. They may require flexible shift patterns or out-of-hours cover within other departments (QC, TS) to permit sampling and aliquotting.
Photo 1:


Operators should carefully consider the requirement for 36-h intermediate hold points. If production samples are taken during normal working hours (typically mid- to late afternoon for downstream operations), then a 36-h time point would require aliquotting in the middle of the night on the appropriate date and may therefore demand additional resources.
The maximum time permitted for sample transfer from production and aliquotting before storage should be defined. That time point is an additional variable that should be controlled to minimize variability and potential impact on product quality. At the end of such time intervals, samples are transferred from production, aliquotted, and either frozen (depending on product stability data) or stored at 2–8 °C for microbial testing (bioburden and endotoxin). Frozen samples are typically analyzed to assess product quality attributes within a defined time period after thawing (e.g., 48 h) to standardize the time and minimize the effect of an additional hold period.
Samples for product stability and microbial control should be included as part of a site sample plan and site data management system. Sample coordination is one of the challenging aspects of these studies, and its importance is usually underestimated (Figure 2). For smooth validation processes, a dedicated person should be accountable for sample tracking and management.
 r;” >
r;” >
Analysis of Intermediate Hold Study Samples
Hold study samples are analyzed for appropriate quality attributes. Any variability observed between time zero (T0) and the maximum hold time evaluated (Tmax) may be assessed. If a variation is greater that the analytical variability defined for an assay (4), then the result is further assessed to determine how hold time affects product quality. Statistical tools could also be employed such as paired t-tests to show no significant statistical difference observed between the T0 and Tmax for each hold point. Scientific judgment should prevail depending on the quantitative values obtained. Additional time points are often held as frozen retains but may be tested to evaluate a trend and define hold time durations. The hold time validated at small scale over three or more consecutive runs may be confirmed by a worst-case run at manufacturing scale — here along with the T0 and Tmax, the drug substance specifications are also reviewed to show that the individual hold times had no impact on the final product quality. To ensure that additional hold periods during sample handling and analysis do not invalidate a study, the maximum time required to complete analytical testing should also be defined as part of the protocol. If a large number of assays is required to support a hold study, then the hold sample may need to be held at 2–8 °C for several days. Although that additional period may still meet the validation criteria for assays, it would extend the hold-point time, so the effect of the time required for analytical testing must be addressed.
Generally a protocol owner and QC department should agree upon a fixed time. The time duration should be long enough to permit completion of all analytical testing required but short enough to limit the effect of this extended hold on product quality. For late-stage products, stress testing or forced degradation data may be available to assess the risk of sample holds required during analysis. One approach to minimize hold times during sample analysis is to aliquot and freeze hold-study samples after the required time. Samples are then tested within a specified time after thawing a sample. In this case, it is important to define and agree on when to start the clock for analytical testing. For example, a hold period could begin as soon as a frozen sample is taken out of a freezer, or it could begin once a sample has been fully thawed and ready for analysis. Some process samples (such as a bioreactor harvest hold) may need extra sample processing to purify product (e.g., protein A chromatography for MAb purification) before analysis (5).Additional sample requirements should be planned well in advance so that the selected time for analysis may be accounted for as part of the agreed testing time. Finally, if possible, all hold study samples from each process step should be thawed and analyzed together to minimize potential influence of both assay and operator variability.
Discussion
In theory, the validation of intermediate hold studies to support late-stage biopharmaceutical production is straightforward. In practice, successful execution requires considerable coordination between quality control, production, and technical groups. Consistent terminology for intermediate hold samples should be considered. An anion-exchange pool may be the same as a cation-exchange load sample, but different terminology within validation protocols, batch records, and sample plans can add complexity during study execution. Maximum time permitted for sample transfer, aliquotting, and testing should also be defined and controlled to minimize variability and potential impact on product quality.
TS, QC, and production groups should clearly define and agree to a time 0 (T0) for each hold point. That time may be as soon as a hold vessel or bag is full — which would include rinse materials during filtration or a UF/DF step — or it could start once a hold bag or vessel is full and has gone through mixing or recirculation to ensure the hold material is homogeneous before sampling. During some process steps (e.g., column chromatography) individual cycles may be collected and stored in separate bags before cycles are pooled, often the following the day. In this case, start of the intermediate pool hold time (T0) is usually defined as the time at which product collection from the first cycle (cycle A) is complete. In such case, eluate from cycle A would spend the longest period at the hold temperature and hence is the worst-case hold time following the process step. Another limitation may be the number of sample ports available within production. This limitation should be evaluated and discussed early so that alternative options for sampling can be identified.
Author Details
At the time of writing, Piranavan Thillaivinayagalingam was senior scientist at the process sciences group of Pfizer Biotechnology Ireland. Corresponding author Anthony R Newcombe is the purification lead at the process sciences group of Pfizer Biotechnology Ireland, Shanbally, Ringaskiddy, Cork, Ireland, 00353 21 500 7856; anthony.newcombe@pfizer.com.
REFERENCES