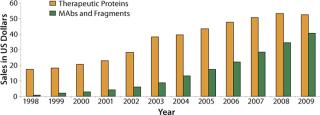
Since the 1980s launch of the first recombinant-DNA–sourced protein insulin, the 1990s introduction of interferons and interleukins, and the first commercial approval of MAbs around the turn of the century, the therapeutic protein market has shown a very healthy growth of 15–19% (Figure 1). Between 1980 and 2004, about 300 antibodies and 400 other recombinant proteins entered clinical trials, totaling about 750 products (1).
A survey of biopharmaceutical production technologies in 2005 shows that Chinese hamster ovary (CHO) cells and a murine myeloma cell line (NS0) among mammalian cells and Escherichia coli (E. coli) among microbial systems remain the tested and most commonly used workhorses, with known safety and productivity profiles and capabilities (2).
More than 66% of approved biopharmaceuticals are glycosylated proteins that require mammalian cell culture. Examples include monoclonal antibodies (MAbs), blood factors, anticoagulants, thrombolytics, EPO, granulocyte-macrophage colony-stimulating factors, specific interferons, hormones, and others such as the Cerezyme, Fabrazyme, Pulmozyme, Myozyme, Naglazyme enzymes. So mammalian cell culture continues to be the cornerstone of production especially for humanized monoclonal antibodies. Over 60% of all new target proteins are produced using CHO cells, and practically all the remainder (<40%) are produced by E. coli. Unpurified antibody titers of 5–10 g/L can be achieved with mammalian cell culture, which is important because clinical doses for therapeutic antibodies are high.
ILLUSTRATION BY CHERYL SCOTT
The narrow focus of applied expression systems is contrasted by a fairly large number of available production methods that could (at least theoretically) be used for heterologous expression of therapeutic and catalytic proteins. The range is broad, and Table 1 summarizes our company’s view of the landscape. Potential methods for producing therapeutic proteins range from prokaryotic bacteria through plant cell culture to mammalian cells and transgenic plants. Comparing the systems in Table 1, our assessment led us to the conclusion that submersed production in a sterile and controlled container (fermentor) with recombinant mammalian cell culture and microbial fermentation will be the preferred method for some time to come.
Table 1: Overview of several manufacturing options for recombinant protein production shows for which technologies Lonza has established expertise and which host cells have a potential — and to what degree (+ + + important, ++ potential, + limited, – not used) — for proteins and/or peptides. In-house columns list cultivation systems Lonza uses for production

Some alternatives to submersed culture may become economically and technically feasible. In 2007 for example, Spök reported that 370 plant-made pharmaceuticals (PMPs) are presently undergoing field trails in the United States and Canada, and about 16 of them are reported to be in clinical trials (3). Furthermore, Sigma Aldrich already uses molecular farming to make avidin, aprotinin, lysozyme lactoferrin, and trypsin at small scales for use as chemicals. Because of strong pressure from the food industry, environmentalists, and consumer organizations, however, a regulatory framework is slow in coming.
Focus on Submersed CultureThe advantage of submersed culture or fermentation is that versatile, state-of-the-art equipment can be used with many different organisms and processes technologies. Continuously stirred tank bioreactors are used in processes ranging from mammalian cell culture to filamentous microorganism fermentation. Combining these manufacturing tools with selected organisms has been tested and considered safe for large-scale manufacturing production of active pharmaceutical ingredients at competitive prices.
Our experience is also in-line with a BioProcess International future. A microbial strain for heterologous protein production can be developed quite rapidly (in four weeks) if a gene is available, and short batch cycles for reader survey carried out in 2007 (4), in which CHO cells were the favorite animal cell line and E. coli the microbial system with the best outlook for the fermentation and product purification (usually about a week) make E. coli and yeasts attractive production organisms.
Although E. coli is the microbial workhorse, there are some 30 other expression systems and microbial hosts available today. The “Microbial Expression” box lists several different Gram-positive and Gram-negative organisms, yeasts, and filamentous fungi that can be used for the heterologous expression of proteins.
One reason so many host and expression combinations exist is certainly the intentional build-up of innovative intellectual property coupled with expected royalty revenues. Another reason is that some proteins and peptides are very difficult to express in established systems, so alternatives are needed. The reason few of these hosts are used regularly by the biopharmaceutical industry is that those have proven suitable for therapeutic protein expression, namely E. coli, Saccharomyces cerevisiae, and Pichia pastoris. Although E. coli is by far the most frequently used host for heterologous protein expression, reports on P. pastoris have increased in recent years.
MICROBIAL EXPRESSION SYSTEMS
Available microbial expression candidates include the following:
Arxula adeninvorans
Aspergillus species
Bacillus brevis
Bacillus megaterium
Candida maltosa
Chrysosporium lu
cknowense
Debaryomyces occidentalis*
Escherichia coli
Fusarium graminearum
Hansenula polymorpha
Kluyveromyces lactis
Kluyveromyces marxianus
Lactococcus lactis
Pichia angusta
Pichia pastoris
Pichia methanolica
Pseudomonas fluorescens
Pseudomonas putida
Pseudozyma flocculosa
Pseudozyma antarctica
Saccharomyces cerevisiae
Schizosaccharomyces pombae
Sordaria macrospore
Staphylococcus carnosus
Streptomyces lividans
Trichoderma reesei
Yarowia lypolitica
* also known as Schwanniomyces occidentalis
WHY P
With a strong preference for respiratory growth, P. pastoris is important to heterologous protein expression for the following reasons:
-
Fast growth up to very high cell densities
-
Regulatory status: P. pastoris is a generally recommended as safe (GRAS) organism; FDA/EMEA approval of the first innovative biopharmaceutical produced by it is expected in 2008.
-
Availability of several signal sequences for protein secretion (e.g., α-factor) and concomitantly simplified downstream processing
-
Integrative expression plasmids are available and single- and multiple-copy integrants with high mitotic stability can be obtained.
-
Possible development of “customized” homogenous glycosylation
-
Widely used with P. pastoris, the AOX promoter is one of the strongest promoters known. Other even stronger promoters can be identified and developed for P. pastoris.
-
P. pastoris can achieve high product titers of secreted proteins (>3 g/L).
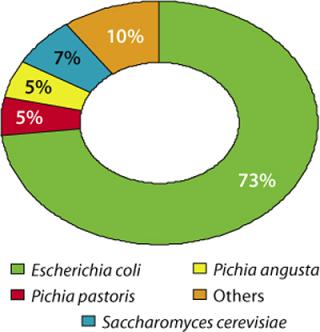
Lonza has worked on 85 microbial expression projects during the past 15 years (preclinical, phase 1–3, and commercial supply projects). Figure 2 illustrates the distribution of requested expression systems based on our analysis of these projects. Among the microbial systems our company offers, we see a clear preference (73 %) for E. coli, followed by 17% of projects using yeasts.
Lonza offers proprietary expression systems for E. coli, P. pastoris, Bacillus subtilis, and Pseudomonas species. The “Why Pichia?” box summarizes why P. pastoris holds a privileged position as our preferred host after E. coli for microbial protein production.
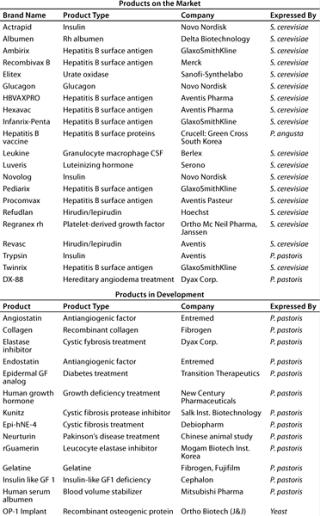
Analysis of therapeutic proteins produced in yeast reveals that for S. cerevisiae a considerable number of well-known, important commercial products have been approved (Table 2). More than 500 products have been reportedly expressed in P. pastoris (5). Most of those are development products. A few important ones, however, are already approved as generics for sale in certain territories (e.g., hepatitis B vaccine) (6). The first innovative biopharmaceutical produced by P. pastoris awaiting FDA approval is the DX 88 enzyme inhibitor from Dyax Corp. of Cambridge, MA (www.dyax.com). The product has orphan drug designation in both the United States and the European Union for treatment of angioedema and fast-track designation in the United States for acute attacks of hereditary angioedema (HAE). FDA approval can be expected sometime in 2008.
Table 2: Therapeutic products expressed in yeast expression systems (adapted from Reference 5)
Another important aspect adding value to P. pastoris is its eukaryotic ability to modify proteins. A number of companies (e.g., GlycoFi, www.glycofi.com; Glycode, www.glycode.fr; and AB Enzymes, www.abenzymes.com) have presented exciting results from optimizing the N-glycosylation of proteins such as IgGs by P. pastoris and S. cerevisiae (7). Given the high cost of mammalian cell culture, those yeast expression systems seem to be a first-choice alternative.
Peptides are another class of products for which there is a significant interest in yeast production. These include human β-defensins and cathelicidin, the two major human antimicrobial peptides in mammalian skin. Several invertebrate, plant, and human defensins as well as other therapeutic peptides have been successfully produced by P. pastoris.
Comparing P. pastoris with E. coli K12The microbial or mammalian strain is the most important ingredient of a bioprocess. It’s not everything, of course; but the most modern facility is worth nothing without a good production strain (8, 9). Table 3 summarizes our company’s view of E. coli and P. pastoris.
Table 3: Comparing E coli and P. pastoris expression systems
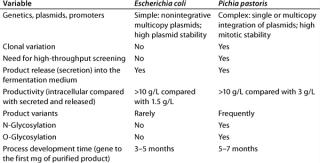
Most modern genetic engineering technologies were developed using E. coli as a model organism. Because the genetics of E. coli are the most understood i
n the microbial world, it is the most prominent production organism for a variety of different therapeutic and catalytic proteins. So Lonza developed its own E. coli expression platform to support strain development programs for its customers. Plasmid-based expression cassettes (e.g., ColE1 and p15A) are used.
By contrast, in P. pastoris all expression constructs must be integrated into the chromosome. Although autonomous multicopy plasmids exist for this yeast, chromosomal integration has been more successful. This more complex integration process is driven by homologous recombination, which may result in clonal variation (10). To identify the most productive clones, more laborious screening procedures are developed and optimized (11). Sometimes thousands of P. pastoris transformants must be analyzed to identify a valuable production clone (12). In our company’s view, such clonal variation requires automated high-throughput screening. Development times — from gene to the first milligram of purified product — are about the same (five to six months) for both E. coli and P. pastoris.
The most obvious advantage of P. pastoris as a production host is its secretion of product into fermentation medium at titers of up to 3 g/L. Primary recovery and product purification are less laborious and time-consuming for this system than for the bacterial one, resulting in more cost-efficient purification processes.
We compared the productivity of E. coli and P. pastoris, and Pichia angusta for an undisclosed peptide precursor (a 30-kD artificial protein) using our company’s well-developed strong rhamnose-inducible protein expression system and a Pichia expression system based on the AOX promoter from Research Corporation Technologies of Tucson, AZ (www.rctech.com). We found similar peak expression levels for secreted concatemer expression in E. coli (periplasmic) and P. pastoris (secreted) at 2–3 g/L. In experiments for intracellular accumulation of the same concatemer in E. coli using a different strong promoter system (pL), we found levels of up to 6 g/L product.
Large-Scale ConcernsP. pastoris has the potential and ability to reliably produce therapeutic and catalytic proteins, to modify protein products after translation, and eventually to reduce costs. An important aspect for cost reduction is the possibility of cultivating P. pastoris at high cell densities and in large fermentors for economies of scale.
Today’s strain performance and process recipes stretch the limits of bioreactor performance. For example, cultivation on pure methanol as used in standard fermentation protocols for methylotrophic yeast (Figure 4) will not deliver desired results. Modern microbial fermentations are carried out at high cell densities, for which high biomass concentrations must be achieved — hundreds of grams of dry cell weight per liter of fermentation broth.
Such conditions can produce inadequate culture conditions, leading to deviations in set goals. Large bioreactors growing E. coli or P. pastoris at high density may exhibit a number of limitations.
Large-scale, high-density fermentation in general (and of methylotrophic yeasts in particular) requires certain measures discussed below. To carry out high–cell-density fermentation using carbon substrates with a high enthalpy of combustion, Lonza has systematically improved process and reactor design details. The most important improvements are in three different areas: medium composition and feeding strategy, reactor design for high cell density, and computer assisted data mining for industrial process improvement.
Medium and Feeding Strategy: P. pastoris was initially developed and used by Phillips Petroleum Company for large-scale production of single-cell protein for animal feed, and it was recognized early that the organism could successfully reach very high cell densities. Unfortunately this yeast tends to intoxicate itself with metabolytes (formaldehyde, formic acid) because its substrate affinity for alcohol oxydases is so high. To avoid such overflow phenomena and reduce problems of heat evolution, Lonza began first to design new media formulations for it based on mixed substrates by testing different sugars and feed regimes in combination with methanol during induction.
An optimal medium composition and feeding strategy was the most important prerequisite for high–cell-density fermentations with high volumetric productivity. We needed to reach constant product quality without degradation products (13) and prevent the production of toxic overflow metabolites during growth on methanol (14, 15).
The example in Figure 3B is a high–cell-density fermentation for production of recombinant avidin using a new mixed-carbon–source medium containing 43% methanol and 57% sorbitol (C-mol/C-mol) with specifically adapted feed strategies (16). These developments had a combined effect of reducing heat evolution as well as oxygen consumption rate and allowing a regular high-volumetric productivity while assuring a stable product quality. Comparison with a high–cell-density fermentation in a feed containing only methanol as the carbon source during the induction phase (Figure 3A) shows clearly that product concentration was increased with the mixed feed. That increased concentration comes from higher biomass yields during mixed substrate growth, with specific recombinant avidin productivity remaining the same as with methanol alone (16). On-line monitoring of the heat production rate by calorimetry (continuous line in both figures) clearly showed that the rate of heat production is decreased ∼40% with a mixed feed, which is very advantageous at large scales.
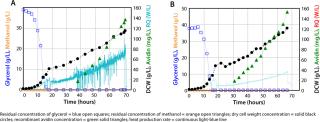
Induction was performed at a constant specific growth rate of 0.03 h−1 during both fermentations because specific growth had a significant influence on glycosylation of the secreted protein. Schenk et al. showed that specific growth rate had an important influence on the number of mannose residues in N-terminal glycans, with a low rate leading to fewer mannose residues (13).
FERMENTATION CRITERIA
Fermentation at manufacturing scales should fulfill these criteria:
-
High fermentation productivity (QP ∼ 50 mg/L/h)
-
A high target in purification yield (50–70%)
-
Consistent product quality (>95%)
-
Reproducible and robust process
-
Short plant occupancy per batch (5–7 days)
-
Low costs for consumables and energy
PROCESS LIMITATIONS
High-density fermentation can involve several serious process limitations that reduce productivity:
-
Oxygen limitation
-
Heat transfer limitation
-
Local nutrient limitations and toxic by products
< /br> -
Product variants and incomplete N-terminal signal sequence processing
-
C-terminal truncation or proteolytic product breakdown
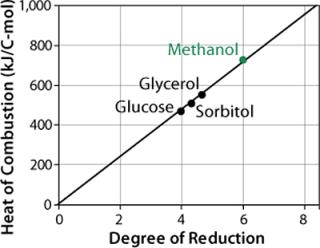
Methanol has a much higher enthalpy of combustion (–727 kJ/C-mol) than glucose, glycerol, or other sugars tested (Figure 4). So replacing part of the methanol with such sugars during induction and product formation considerably reduces heat production as well as the risk of cellular intoxication with overflow products.
Reactor Design for High Cell Density: Once the medium was adapted, high oxygen transfer rates (OTRs) had to be assured, mainly through appropriate reactor design and overhead pressure. High–cell-density fermentations need significantly higher OTRs than those needed in, for example, secondary metabolite fermentations. Standard large-scale fermentors typically reach OTRs of ∼150 mmol/L/h using air and normal pressure for aeration. Lonza has designed and built 15- to 75-m3 bioreactors to reach OTR values of over 400 mmol/L/h. They were specifically designed for high–cell-density cultivation of microbial cells. Moreover, we can increase the partial pressure of oxygen (pO2) by mixing pure oxygen to the air stream entering a fermentor, which further increases its OTR. Foaming is another problem that can arise at high cell densities and high aeration rates, but it is not discussed here. Lonza integrated foam disruption and aeration by testing the effectiveness of a range and combination of impellers for simultaneous stirring and foam disruption (18).
One crucial point often becomes a limiting factor for microbial high-density fermentations: OTRs limited by heat removal. Heat evolution typically remains the critical and limiting factor. This is a consequence of the thermodynamics of microbial activity. The ratio of volume to surface area increases with larger volumes, so the amount of microbial heat present often exceeds the heat transfer and heat removal capacity of large fermentors.
About 40–50% of available enthalpy (through anabolism during growth) in a microbial substrate is conserved in the biomass, and the remaining is given off as heat. The formula below shows that microbial heat formation (QW) expressed in kcal/h depends on the working or liquid volume of a bioreactor (VL) as well as growth rate (µ), biomass (X), and microbial heat formation expressed in grams of biomass per kcal. This formula shows that if as much as half of the available enthalpy in an oxidized substrate is given off as heat, enormous quantities of kilocalories must be removed:
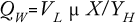
P. pastoris has an additional advantage of achieving very high cell densities in fed-batch culture while “wasting” a lower fraction of substrate than does E. coli, for example. The maintenance energy of P. pastoris is lower than that of other microorganisms (17, 19). Nevertheless, heat evolution does set clear limits for this yeast as well.
The cooling capacity of larger fermentors (e.g., 50 m3) removes 10–15 kW per cubic meter of liquid volume. Design features for high cell densities as shown in Figure 5 allow removal of >40 kW/m3, but as much as 80 kW/m3 may need to be removed during a high-density fermentation. Because an infinite number of cooling baffles or coils cannot be installed, the temperature difference (ΔT) between cooling liquid and fermentation broth must be increased. That usually involves using brine or replacing actual cooling liquids with N2 or direct N2 injection. But that temperature difference cannot be too large because ice formation at the gas–liquid interface can present further problems.

However, using liquid N2 can create a need for extensive fermentor adaptations as well as truckloads of compressed gas each day. Direct injection of it into culture medium can strip out dissolved oxygen, as well. Consequently, liquid N2 may be justified for indirect cooling only in cases of severe limitations due to fermentor geometry.
So indirect cooling of culture medium during fermentation by a jacket and coils remains the standard procedure. For indirect cooling applications, however, liquid N2 can be used either to cool or replace the heat transfer fluid. For operating temperatures ∼30 °C, cooling water and glycol or ammonia chillers are all typically chosen before opting for a nitrogen-based system because of their lower cost.
As mentioned, use of mixed feeds during the induction phase of P. pastoris processes can significantly diminish heat production rate. In combination with state-of-the-art fermentation equipment, this strategy allows high–cell-density fermentation for high and reliable recombinant protein productivity.
Data Management: Huge amounts of on-line and off-line data are typically logged and stored during routine manufacturing process monitoring. We developed a process that supports analysis and interpretation of manufacturing databases for industrial high–cell-density processes at 50-m3 scale with >90 primary variables logged and ∼100,000 time series of original and derived signals available after processing (20).
Our generic statistical methodology serves as a flanking measure to control fermentations because it allows indirect monitoring of important data that are inaccessible in real time (e.g., product concentration), as well as identification of factors that affect process variation and extraction of trends, patterns, and correlations. Because no mathematical models were readily available for these tasks, as is often the case, we applied data-driven, computer-intensive statistical methods with conditional graphics (e.g., spline regression, bootstrap confidence interval estimation, median polishing, and wave-shrink smoothing). Data collection, processing, and analysis (Figure 6) are indispensable parts of optim
izing production processes as well as valuable tools for management decision-making.
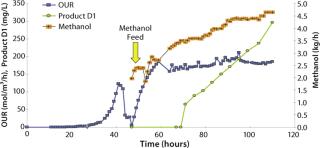
Productivity, protein secretion, and product quality are the three major issues to consider when selecting a microbial production host. In a first example, we present a pilot fermentation at 1.5-m3 scale for production of an undisclosed therapeutic peptide that was released into the fermentation medium up to a peak titer of 0.3 g/L. Product expression driven by the AOX promoter was induced by methanol. Batch phase growth was on glycerol, and induction-phase growth was on pure methanol. A robust, reliable two-phase fed-batch fermentation regime was used. Figure 7 shows oxygen uptake rates during both phases as well as the methanol feed rate and accumulation of product in the fermentation medium.
P. pastoris processes sometimes exhibit N- and C-terminal product variants that usually represent the major product impurities in fermentation supernatant. Because of their nearly identical physicochemical properties, sophisticated purification is needed to eliminate them. Product variants result from incomplete or incorrect N-terminal processing and C-terminal truncation by proteolytic cleavage. The secretory pathway in P. pastoris requires Kex2, a typical eukaryotic protein processing protease. It cleaves the signal sequence at a dibasic recognition site and shows high selectivity for positively charged residues at P2 and an even more stringent requirement for Arg at P1 (21). Depending on the amino-acid sequence of a given protein, Kex2 recognition sites might be mimicked within it, leading to N-terminally modified product variants (22). In many cases, such product variants can be prevented by careful fermentation optimization or use of protease-deficient mutants.
In another example, where a cytokine was produced, product-variant generation could not be prevented. Process development and CGMP production at the 1,000-L CGMP scale was performed using P. pastoris (RCT strain and AOX-driven with methanol-induced expression). In this case, preparative HPLC was used to purify the cytokine to a therapeutic level >95%.
P. pastoris (and P. angusta) can be developed as reliable workhorses for heterologous protein production. Compared with E. coli, which is by far the most frequently used microbial host, the Pichia expression system is not yet fully developed. Generation of product variants and undesired O-glycosylation must be tightly controlled by strain development and specific fermentation protocols. Moreover, the need of high–cell-density fermentation using methanol as an inducer typically causes excessive heat production. Nevertheless, despite these drawbacks, we are convinced that Pichia species have great potential as a manufacturing organism for proteins.
Our reasons are several-fold. First, there is opportunity for the reduction of cost of goods (COGS) because product is released into the fermentation medium, which considerably simplifies downstream processing. Fermentation procedures can be developed to maximize productivity in high–cell-density fermentations, and Lonza has developed mixed-feed fermentation protocols for methanol induction. On top of that, we have developed an alternative Pichia expression platform that does not use methanol. Most importantly, however, Pichia species can perform N-glycosylation that can be engineered to produce homogenous and customized glycosylations.
REFERENCES
am Disruption”. Biochem. Eng. J. 10:182-195.