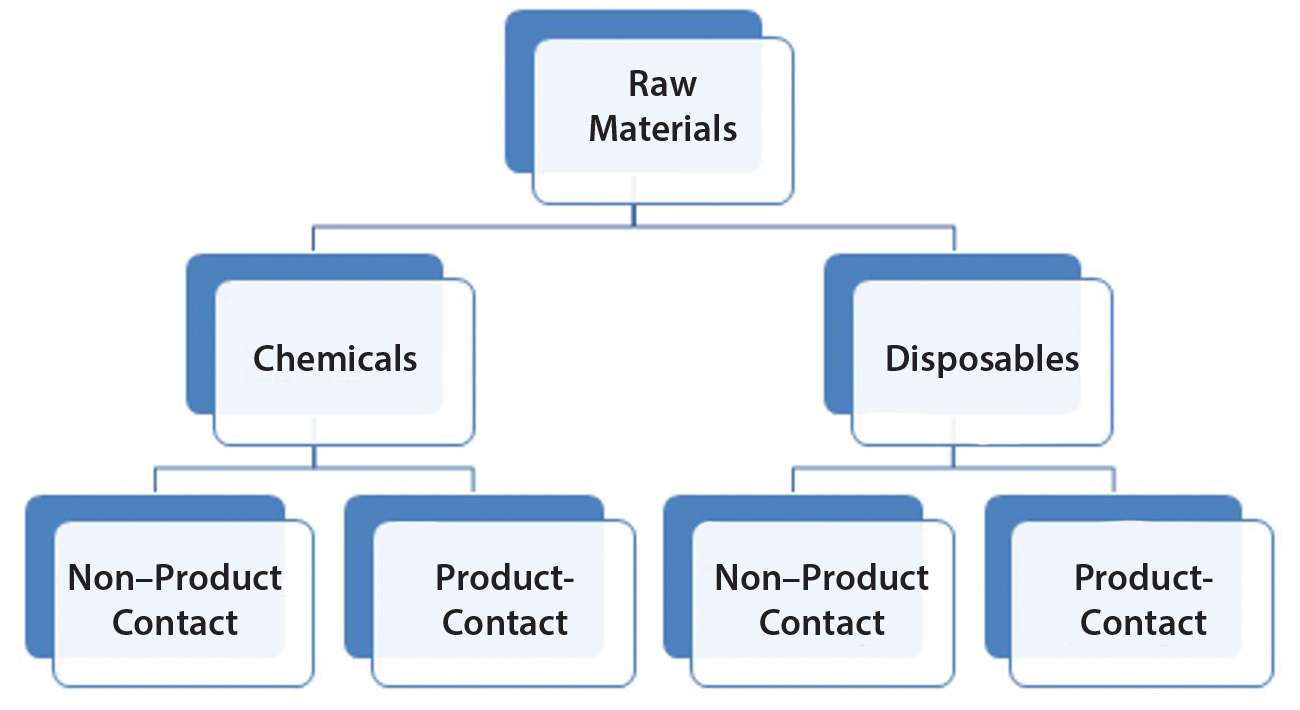
Figure 1: Raw materials high-level classification
Ensuring a continuous supply of safe medicines to patients is a key objective for both health authorities and the pharmaceutical industry. A critical component to that end is maintaining a reliable supply of qualified raw materials (RMs). Manufacturers must ensure not only the suitability of RMs for their intended use in a manufacturing process, but also their highest attainable safety with regards to viruses and other adventitious agents. The need to apply a risk-based RM control strategy is in line with regulatory expectations and is recognized within the biomanufacturing industry (1–3).
Viral contamination is a potential safety threat common to all animal- and human-derived biologics produced in mammalian cell culture (4). A viral contamination can arise from a contaminated cell line source or from adventitious introduction of virus during production. Although contamination of bioreactors and biologic therapeutics is rare, several manufacturers have reported such incidents in biotechnology manufacturing processes or final products in the past decades (5, 6), and RMs often were the primary suspected cause (7–9). A contaminated final product could have dire patient consequences. Manufacturers can spend a lot of time decontaminating affected areas and performing other activities needed to resume commercial production following a contamination event. A lengthy downtime or shutdown not only affects drug manufacturers, but also could affect patients (e.g., in case of a product stockout) (10–12). Therefore, efforts toward preventing such contamination events are essential. Those efforts should address the growing complexity and diversity of supply sites, processes, and products. A first step to that end is identifying and prioritizing potential causes through risk analysis (e.g., by conducting a failure modes and effects analysis, FMEA). In a perspective paper, the US Food and Drug Administration (FDA) recommended a systematic risk assessment, risk management, and response program to ensure that appropriate measures and controls are in place to ensure patient safety (13).
The ICH Q5A guideline serves as the primary viral safety guideline for biologic manufacturing processes based on animal and human cell cultures (14). Regulatory guidelines and guidance documents are written to ensure viral safety of biotechnology products through the application of an effective virus testing program (14), which can be achieved using a three-tiered approach:
- selecting and testing of cell lines and other raw materials
- assessing capacity for viral clearance and inactivation by a manufacturing process
- in-process and/or final-product virus testing.
Yet because regulatory guidelines typically delineate principles to consider (in the case of ICH Q5A when assessing viral safety), the burden is on biotechnology manufacturers to select and implement the most up-to-date solutions — both technologies and mitigation approaches — that are best suited to their current specific processes. The need to address those challenges has facilitated a shift from cell-based to nucleic acid–based adventitious agent detection technologies (15).
Scope
Herein we focus on virus contamination risk assessment as it pertains to RMs for production of a drug substance (DS). We review potential risks during upstream (cell culture) and downstream (isolation and purification) operations. Although out of the scope of this discussion, drug product (DP) generation operations (e.g., fill–finish) also must be assessed.
Our case here assumes use of an ICH Q5A–compliant system. It entails a bioreactor-cultivated, mammalian-production system (e.g., Chinese hamster ovary, CHO, cells) using RMs derived from nonanimal (and nonhuman) sources (or if any animal-derived RMs are used, their source and viral testing are well documented). The downstream process contains multiple independent viral clearance (e.g., through filtration and chromatography steps) and inactivation mechanisms (e.g., exposure of process intermediates to virus inactivating chemicals).
Utilities-related systems (e.g., airflow, gasses, water, and other fluids), chemical solutions preparation, and operations-related virus risk assessments and validation studies are outside of the scope of this discussion because they are specific to company sites and should be evaluated at each site.
We do not discuss production host-cell–source virus risk assessments, process-step viral clearance, and virus inactivation studies (and any other studies conducted in compliance with applicable regulations). Such studies are considered standard for all biopharmaceutical products and are performed independently by biopharmaceutical companies. Yet the aforementioned studies would serve as important reference documents and should be cited as appropriate in RM virus risk evaluations, as discussed below.
Approach
In drug manufacturing, all RMs (which companies typically list in a bill of materials, BoM) entering a process should be evaluated for viral safety. Manufacturers can use inventory management software (e.g., from SAP) of the BoM for DS production to gather all RMs for comprehensive analyses. Standard operating procedures (SOPs), warehouse orders, and technology transfer lists (if applicable) also should be reviewed for complete RM coverage. Every RM is evaluated, assigned a risk level, and (if needed) prioritized for further mitigation. Legacy commercial processes (which may have been established before current standards were put in place) also should be evaluated to ensure that they meet current good manufacturing practice (CGMP) and current regulatory expectations. If they do not, additional mitigations should be placed as needed.
RMs used in the bioproduction of a DS are divided into two general groups termed here as chemicals and disposables (Figure 1). That is to reflect inherent risk-level differences as a potential source of viral contamination and that the latter (disposables) also can be regarded as process aids. Both disposables and chemicals (regardless of their nature and mode of use in a process) are further categorized based on whether they have product contact. Product contact would significantly increase risk levels. Mitigations (for viral risk) in place should be evaluated to confirm adequacy, commensurate with the level of risk (or identify any potential gaps) and the body of supporting documents is to be indexed for reference.
Assessment
RMs for biomanufacturing include all process inputs and production cell sources. Examples include chemicals (such as those found in media, buffers, resins, excipients, process agents, and cleaning solutions), disposable systems, and packaging. RMs are procured from a wide range of qualified suppliers and manufacturers, and they undergo different testing programs (e.g., assays). Thus, variations are both inherent (due to a broad range of RM sources) and a result of practical properties (e.g., type of RM, how it’s stored and tested, and where it is used in a process) and are considered in evaluation processes.
The first part of our assessment focuses on chemicals that present a higher potential virus contamination risk relative to disposable RMs. The second part focuses on disposables (nonchemicals) such as bags, tubing assemblies, filters, and disposable supplies. Both assessments take into consideration whether the RMs have direct or indirect product contact and their proximity to the final product. As noted above, all remaining RMs also should be evaluated for viral risk (e.g., cell bank and RMs used for cell-bank generation, solutions, buffers, calibration standards, testing solutions, and aids that are handled within cleanrooms and manufacturing areas).
Chemicals: Notwithstanding the types of materials, where in a process they are being used, and existing mitigation measures, materials defined as chemicals (powders and solutions) inherently present a higher risk than other RMs (defined as disposables) for a DS process. So chemicals are scrutinized to a higher degree.
Chemicals may be evaluated for three aspects: where used (proximity to drug product or patient), nature of chemical (its production process and assigned grade), and quality oversight (certified level of testing, supply chain complexity, and visibility).
The where used factor in a process-step area includes evaluating whether a material is used for upstream processes (USPs), downstream processes (DSPs), excipients, or formulations.
Nature of chemical includes knowing whether a material contains animal-derived components or is an animal-component–free (ACF) material and whether viral inactivating steps were used in manufacturing a given chemical.
Quality oversight includes
- viral testing, vendor certificate of analysis (C of A), and in-house testing
- supply chain and level of control, oversight, and transparency over production through shipment and arrival on site (e.g., routine audit schedule and per inspection plan, including vendor and in-house viral testing and container integrity check)
- supporting documentation (reference to any existing studies, measures, and verifications) from suppliers and internal (e.g., development reports and SOPs).

Table 1: Chemical raw material (RM) categorization (ACF = animal-component free)
Table 1 shows a chemicals’ assessment based on the aforementioned criteria. The assessment starts with sorting chemicals into the three areas of use in a production process: upstream, downstream, and excipient/formulation. Risk level would increase in that order, respectively. Note that some chemicals can be used in more than one area. In such cases, the chemical is assessed according to the area that is closer to a drug product (among common areas in which it is used) to receive the highest potential risk evaluation. Whereas an excipient would have contact with a drug product, if the chemical is used in upstream or downstream production, contact with drug substance is determined. Using RMs that are ACF and viral inactivation steps in the RM’s production process lowers the contamination risk compared with meeting only one of those two criteria. Otherwise, if the risk is not low, you should reevaluate the “where used” factor, taking into consideration that viruses that might be entering a downstream process generally don’t have a potential amplification host and medium (unlike cultured expression cells used in upstream processes that might be capable of virus propagation). Finally, the level of quality oversight is determined and references are made to pertinent supporting documents (e.g., C of A, incoming testing results, and audit reports). The evaluation approach presented here also takes into consideration all relevant studies performed within the company, and references should be made to pertinent reports, including risk assessments and available publications in the public domain.
The above aspects can be ranked (including relative weighing) by assigning a numerical value (e.g., a risk priority number, RPN, in an FMEA) or by using a qualitative strategy. The same principles are applied in justifying the assigned risk level, whether numerical or quantitative. Each chemical category is ultimately assigned with a risk value of high (H), medium (M), or low (L). A mitigation approach of “virus testing,” “process-step viral-load reduction,” or “chemical not conductive to viral propagation” can be assigned accordingly.
In a case study using this approach, we evaluated chemicals in terms of type/nature and process use of RMs through the type of mitigations already in place and through reviewing the existing body of supporting work identified. We separated and categorized RMs based on the use during the production process. The chemical use categories consisted of media (M), cleaning/stripping upstream (CU), cleaning/stripping downstream (CD), buffer downstream (BD), buffer upstream (BU), formulation upstream (FU), formulation downstream (FD), column resin (C), and working cell bank (WCB) storage (W). The bottom row in Table 1 provides an example evaluation of one chemical (cupric sulfate) out of the entire chemical RMs evaluated for a legacy process (internal data, not shown) according to the various aspects discussed.
Each chemical is assessed based on the criteria listed in Table 1. Ideally, all chemicals would be considered ACF. In addition, chemicals such as media powder would be tested in-house for potential viral contamination. For other chemicals such as cell culture medium and medium components, a vendor performs adventitious viral testing (AVA) and reports those results in a C of A.
Some chemicals are evaluated based on existing studies and publications, which should indicate whether these components do not support viral propagation. Some chemicals are exploited for their known viral inactivating properties, including Triton X100 (17), polysorbate 80/TNBP (18, 19), and cupric sulfate — the chemical given as an example in Table 1 (16) — which all have virus-inactivating properties.
Manufacturing processes for RM components (including secondary suppliers) also should be evaluated. This information should be requested from primary suppliers because the specific sources and process types (e.g., synthetic, crystallization, plant-derived, and bacterial fermentation) are important factors in risk assessment. Manufacturers also would need to know whether process steps for RM production are virus-inhibitory (such as low pH, high temperature, various concentration, chromatographic and filtration steps, and use of organic solvents). For example, the manufacturing process of some amino acids often entails a process that is not conducive to viral growth or that is inhibitory to viral growth (e.g., serine and glutamine synthesis).
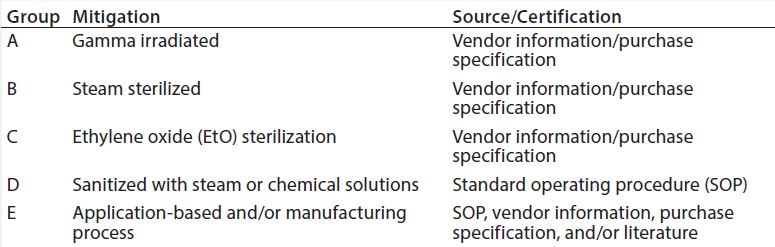
Table 2: Disposables grouping according to the mitigation used
Disposable components required to produce a drug substance are divided into group (Table 2) according to their production process, treatment (19) before use (irradiation, chemical,or heat treatment, Table 3), or mode of use in a drug substance production process. Each disposable RM with direct or indirect product contact is evaluated individually to determine its risk of introducing viral contamination. Although this evaluation focuses on using different specific mitigating treatments, disposable RMs are assessed using the same three criteria as for chemical RMs (where used, nature of RM, and quality oversight). RMs that are not treated (sterilized or sanitized, Table 3) are further evaluated on their “purpose of use” (e.g., whether they come into contact with a product during sample handling). A risk level is thus assigned to all RMs and a justification provided in a format similar to that presented for chemicals in Table 1.
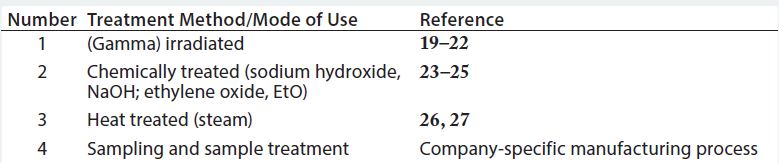
Table 3: Evaluation of disposables based on how they are produced and/or treated before use and based on their mode of use in the drug manufacturing process
In this approach, if disposable RMs used in drug substance production processes are processed or treated (by irradiation, chemicals, or heat) or if they are used solely for sample handling, they are deemed to present low to no risk as a potential source of viral contamination.
In a case study following this approach (e.g., for tubing assemblies and sample bags; data not shown), each disposable RM was assessed based on the criteria above. Most disposables used in a drug substance manufacturing process fell into one or more categories listed in Table 2 in groups A–D, classifying them as “low” risk as a source of viral contamination. Few disposables in the drug substance production process were neither sterilized nor sanitized before use (group E). Those materials were evaluated to pose a low viral risk on the basis of their application and/or manufacturing-process of the RM (e.g., high temperature or pressure treatment of resins or cleanroom manufacturing). For example, tubing assemblies used for Triton X-100 transfer pose a low risk because Triton X-100 (itself) is a virus inactivating agent (17) and because these assemblies are manufactured in a cleanroom environment (with the purchase specification serving as a certification source). Pipet tips for sampling serve as another example of disposable RMs justified on the basis of its use (for sampling); they are not returned to product streams with the source/certification being the internal SOP.
Some literature has reported cases of viral contamination, with the likely sources being chemical RMs (28). Disposable and nonchemical RMs are therefore inherently lower risks for viral contamination as discussed above, and virus risk is mitigated by multiple avenues.
Evaluating All Risk Factors
A key principle in the approach presented is to match mitigation stringency with the inherent risk of a given RM. Disposables (e.g., bags, tubing assemblies, filters, and other single or multiuse disposable supplies) are addressed separately from chemicals (all nondisposable RMs) because they inherently present a lower risk by the nature of their production process and/or treatment (e.g., being irradiated or autoclaved). Within the chemicals group, preparation of chemical solutions, buffers, and in-house process intermediates can be addressed separately (because such operations are often site dependent).
All drug-production process materials are reviewed to identify and categorize the chemicals into three areas of a production process: upstream, downstream, and excipient/formulation. Each chemical RM is evaluated individually to assess the risk of its introducing a viral contamination or as a potential breach that might enable a viral contamination. That evaluation takes into consideration whether an RM is animal derived or made in a process that makes use of animal-sourced components, where it’s used in a process (upstream, downstream, or as an excipient), whether it contacts a product, whether there are existing viral testing in place, and other factors.
Chemicals identified as potential medium/high risks should be further mitigated. This process can include additional measures in a supply chain (e.g., better packaging procedures), routine virus testing, or even replacing a high-risk component with a lower-risk one (e.g., replacing an animal-derived component with a synthetic equivalent). If as currently is the case for many modern processes, no chemical RMs are animal derived, then their risk as sources of viral contamination is significantly reduced. Following needed mitigations, all chemicals listed in a BoM should be considered to be of low viral risk. The evaluation should refer to existing pertinent supporting body of work, including risk assessments, reports, and associated studies (e.g., viral clearance studies and facility and operations review).
Likewise, all materials for drug-substance manufacturing should be reviewed to identify and categorize disposable RMs. Each disposable RM with direct or indirect product contact is evaluated individually to assess its risk of introducing a viral contamination. Disposables with no product contact pose minimal risk and can be excluded from the assessment.
Some disposable RMs in a manufacturing process are neither sterilized nor sanitized before use (which can be verified, for example, by reviewing purchase specifications, vendor certifications, and company SOPs). For such materials, mitigations arising from their manufacturing processes (e.g., high-temperature and high-pressure processing steps or cleanroom manufacturing) also should be considered. The mode of use in a drug-substance manufacturing process should be taken into account for such materials. For example, a disposable that is used solely for sample treatment poses a low viral risk, even if it is not sterilized or sanitized before use (because the treated sample is not returned into the process stream).
Upon performing that evaluation, manufacturers can conclude that all RMs (disposables and chemicals) used in a drug-substance manufacturing process present a low risk for viral contamination. Some scenarios can occur in which a low risk level cannot be assigned to a certain RM (e.g., in the transfer of a legacy process that was developed before a time when an assessment to standards such as those discussed here was expected before clinical manufacturing). In such a case, additional measures must be implemented to reach that goal.
For disposables, the evaluation can be based on their treatment before use (e.g., gamma irradiation, heat or chemical treatment), their manufacturing process, or their mode of use (e.g., application without product contact).
For chemicals, the evaluation can be based on whether an RM is animal-component–free (ACF), whether in-house testing is in place, and on a chemical’s viral growth-promotion capabilities. The manufacturing process of some RMs can be inhibitory to viral growth, so this factor also should be considered.
Appropriately conducting an RM viral-risk assessment and documenting results and conclusions using quality-system procedures from a site-specific biopharmaceutical company would serve as important authorizations and justifications for procuring and using RM components in commercial production.
Acknowledgments
For their contributions to the approach presented herein, we thank the following colleagues: Shawn Liu, Sandra Sternberg, Alexander Poguntke, Frank Dittmer, and Scott Probst. We are also grateful to Anita Bawa, Petra Wippich-Quadt, and Jay Benson for their sponsorship and support and to Garrison Beye for reviewing the manuscript.
References
1 Guidance for Industry: Q8, Q9, and Q10 Questions and Answers — Appendix Q&As from Training Sessions. US Food and Drug Administration: Rockville, MD, July 2012; www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm313094.pdf.
2 Khan AS, et al. PDA/FDA Adventitious Agents and Novel Cell Substrates: Emerging Technologies and New Challenges, 3–4 Nov. 2011, Rockville, MD. Conference Report. PDA J. Pharm. Sci. Technol. 66(6) 2012: 502–511. doi:10.5731/pdajpst.2012.00895.
3 Shimoni Y, et al. A Risk-Based Approach to Supplier and Raw Materials Management. BioProcess Int. 13(10) 2015: 10–15.
4 Merten OW. Virus Contaminations of Cell Cultures: A Biotechnological View. Cytotechnol. 39(2) 2002: 91–116. doi: 10.1023/A:1022969101804.
5 Wiebe ME. Virus Contamination of Cell Culture Manufacturing Operations: A Question of When, Not If. WCC PDA, San Francisco, June 17, 2010.
6 Liu S. Effective Prevention of Bioreactor from Viral Contamination. IBC-Biologics Development and Production, Laguna Beach, CA, 24–28 Feb. 2013.
7 Garnick, RL. Experience with Viral Contamination in Cell Culture. Dev. Biol. Stand. 88, 1996: 49–56.
8 Garnick RL. Raw Materials as a Source of Contamination in Large-Scale Cell Culture. Dev. Biol. Stand. 93, 1998: 21–29.
9 Nims RW et al. Detection of Cache Valley Virus in Biologics Manufactured in CHO Cells. BioPharm Int. 21(10) 2008: 89–94.
10 Skrine J. A Biotech Production Facility Contamination Case Study: Minute Mouse Virus. PDA J. Pharm. Sci. Technol. 65(6) 2011: 599–611. doi: 10.5731/pdajpst.2011.00823.
11 Jones N. Identification and Remediation of a Cell Culture Virus Contamination. PDA J. Pharm. Sci. Technol. 65(6) 2011: 615. doi: 10.5731/pdajpst.2011.00822.
12 Geigert J. The Challenge of CMC Regulatory Compliance for Biopharmaceuticals and Other Biologics, Second Edition. Springer Science: New York, NY, 2013: 68-85.
13 Xu L et al. Role of Risk Assessments in Viral Safety: An FDA Perspective. PDA J. Pharm. Sci. Technol. 68(1) 2014: 6-10. doi: 10.5731/pdajpst.2014.00959.
14 Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin. International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Q5A(R1): 1999.
15 Kljavin et al. Adventitious Agent Testing of Biologicals: Changing to a New Frontier of Technology, Cell-based to Nucleic Acid-Based Detection. State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization, Volume 3. 2015: 227–244. ISBN: 13: 9780841230316. eBook ISBN: 9780841230323.
16 Borkow G, Gabbay J. Copper As a Biocidal Tool. Curr. Med. Chem. 12(18) 2005: 2163–2175.
17 Roberts PL.Virus Inactivation by Solvent/Detergent Treatment Using Triton X-100 in a High Purity Factor VIII. Biologicals 36(5) 2008: 330–335.
18 Seitz H et al. Comparable Virus Inactivation by Bovine- or Vegetable-Derived Tween 80 During Solvent/Detergent Treatment. Biologicals 30(3) 2002: 197–205.
19 Guidelines on Viral Inactivation and Removal Procedures Intended to Assure the Viral Safety of Human Blood Plasma Products. WHO-TRS-924_A4-1: 2004.
20 Gauvin G, Nims R. Gamma-Irradiation of Serum for the Inactivation of Adventitious Contaminants. PDA J. Pharm. Sci. Technol. 64(5) 2010: 432–435.
21 Moore M. Inactivation of Enveloped and Nonenveloped Viruses on Seeded Human Tissues by Gamma Irradiation. Cell Tissue Bank 13(3) 2012: 401–407. doi: 10.1007/s10561-011-9266-0.
22 Sullivan R, et al. Inactivation of Thirty Viruses by Gamma Radiation. Applied Microbiology 22(1) 1971: 61–65.
23 Jeong EK, et al. Inactivation of Influenza A Virus H1N1 by Disinfection Process. Am. J. Infect. Control 38(5) 2010: 354–360. doi: 10.1016/j.ajic.2010.03.003.
24 Use of Sodium Hydroxide for Cleaning and Sanitizing Chromatography Media and Systems. GE HealthCare Application Note 18–1124-57AG.
25 Rutala WA, et al. CDC Guideline for Disinfection and Sterilization in Healthcare Facilities. 2008; www.cdc.gov/hicpac/pdf/guidelines/Disinfection_Nov_2008.pdf.
26 Lelie PN, et al. Inactivation of 12 Viruses by Heating Steps Applied during Manufacture of a Hepatitis B Vaccine. J. Med. Virol. 23(3) 1987: 297–301.
27 Schleh M, et al. Susceptibility of Mouse Minute Virus to Inactivation By Heat in Two Cell Culture Media Types. Biotechnol. Prog. 25(3) 2009: 854–860.
28 Wisher M. Virus Risk Mitigation for Raw Materials, A European Perspective. BioProcess Int. 11(9) 2013: 12–15.
Corresponding author Yuval Shimoni is principal engineer, Venkatesh Srinivasan is director of manufacturing sciences, and Robert To is senior quality control manager at Bayer LLC, Berkeley, CA; 1-510-705-5775; yuval.shimoni@bayer.com. Andre Pastor is a technology expert at Bayer Technology Services GmbH, and Joerg Peters is head of manufacturing sciences at Bayer in Wuppertal, Germany.