Elements of the biopharmaceutical industry’s new operating paradigm have inevitably created an immediate need to condense, interpret, and relate their implications to existing regulatory and industry practices. This also provides us with an opportunity to look at them in a broader context and in relationship to one another. Such a perspective may open up new directions in discussion on how design and control aspects of biopharmaceutical manufacturing are likely to evolve. These are exciting times for scientists, engineers, statisticians, quality professionals, and regulators alike because contributions from each are needed to establish new practices that will deliver on the intentions and expectations of these new initiatives.
In Chapters 2 and 3, we explore in a harmonized context the fundamental concepts of quality by design (QbD) (1, 2), process analytical technology (PAT) (3), quality risk management (QRM) (4) and pharmaceutical quality (5,–6) initiatives, as well as the emerging field of systems biology (SB) (7). Particular emphasis is placed on the future direction of process design and control activities for biopharmaceutical manufacturing processes.
Continual or Continuous Improvement
Some authors prefer the term continual improvement over continuous improvement. Although both are similar, they may invoke different meanings within a certain context. We chose to use the first term throughout these chapters for consistency.
Modern biopharmaceutical development and manufacturing activities are inherently complex, so the distinct path that they follow is often highly iterative and interactive. It proceeds from identification of a target product profile through product and process development to process scale-up and technology transfer, and finally ending in manufacturing implementation that produces a commercial product.

BIOVITRUM (WWW.BIOVITRUM.COM)
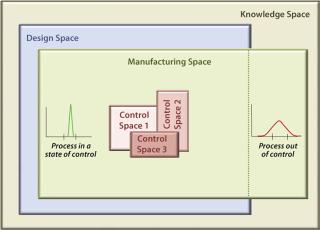
Figure 1 models a biopharmaceutical development and manufacturing path according to the principles of QbD and illustrates the role of QRM activities. QbD encompasses a scientific, risk-based, holistic, and proactive approach to pharmaceutical development. It constitutes a formal design effort from product conception (at definition of a target product profile) through commercialization to achieve full understanding of how process parameters and product attributes relate to process and product performance. This includes quality and clinical outcomes as defined in terms of safety and efficacy. Figure 2 further illustrates the concepts of QbD implementation as a hierarchy and relationship between the individual “spaces” required to achieve a manufacturing implementation that is in a state of control. The knowledge space here is the sum of existing information derived from prior knowledge and the body of scientific data about product and process (8). This provides a basis for delineating the design, manufacturing, and control subset spaces.
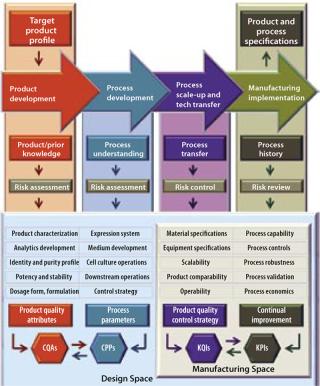
The Design Space Concept: The model introduces the design space, portrayed here in a narrow sense so it can be distinguished from the manufacturing space, although in most cases the term covers all aspects of product/process design and performance. Design space harbors a number of distinct design activities intended to establish product quality attributes (particularly those that impart clinical performance) and subsequent process parameters (emphasizing those critical to quality), respectively referred to as critical quality attributes (CQAs) and critical process parameters (CPPs). Their causal linkage (functional relationship, a primary element in process design) must be characterized during the design phase — a regulatory expectation. Individual CQAs are generally established for each drug substance, its excipients, the drug product, and even related intermediates.
A design space is briefly defined as a multidimensional combination and interaction of process input variables (material attributes) and process parameters that have been demonstrated to provide assurance of quality (1). Consequently, it is derived from values pertaining to materials, procedures, and equipment used; and to product quality attributes, process parameters, and environmental conditions. The design space can be more specifically described as the intersection of quality attributes and process parameters that defines a multidimensional response surface linking elements of both. Its establishment is based on the product knowledge and process understanding of multidisciplinary teams. A design space can be conceived for one manufacturing process established for a particular product or expanded and applied across a class of molecules and platforms, giving rise to what some have dubbed a “super design space.”
Design space is established through a living document. It should be reviewed periodically as part of the related quality system and updated as new understanding is gained through such manufacturing experiences as
-
discovery of new control parameters
-
critical knowledge gained from failures and investigational reports
-
operational adjustments within the design space.
It may also be expanded based on results from further experimentation or through additional knowledge acquired, for example, to propose a reduction of the number of CPPs and CQAs, modify specifications, or eliminate certain in-process tests —although these require renewed FDA approval. Shrinking a design space because a process is less robust than initially anticipated represents a dilemma for regulatory reporting because design space is determined by process capability (among o
ther factors), which itself determines process robustness (see online appendix).
The Manufacturing Space Concept: Parallel to the idea of design space, Figure 1 also depicts a manufacturing space that promises to further enhance product knowledge and process understanding. (We reintroduce the term here to segregate the actual technology transfer and scale-up activities and manufacturing implementation from design activities conducted in the laboratory.) The enhancement is essentially accomplished by correlating individual product and process outputs, such as key quality indicators (KQIs) and key performance indicators (KPIs), to their respective underlying design elements in the design space. Examples of KPIs include stock turns, on-time in full (OTIF), right first time (RFT), process capability, overall equipment effectiveness (OEE), and cycle time. The concept of KQIs is introduced here to provide a metric for quality control strategy during process scale-up and technology transfer.
The concept of a manufacturing space is a valuable tool to capture and correlate results from process scale-up and technology transfer activities as well as manufacturing implementation. An important principle in the development of a manufacturing space is the engineering tool known as scale correlations. Here they are used to refer to actual scale-up from laboratory to pilot and commercial scale — or for the modeling of manufacturing-scale processes in scaled-down versions for process development and validation in a design space. Because scale correlations are derived from a mixture of experimental and mathematical operations, they present a semiempirical approach to translating operating conditions between different scales or pieces of process equipment.
By virtue of the often highly iterative nature of development and manufacturing efforts, formal links must be established between the design space and manufacturing space. This must be done while safeguarding the overall progression of the development path to commercial product, because time remains the critical driver.
The Control Space Concept: A proven acceptable range (PAR) of operation for each CPP is established for process control and firmly embedded within the design space. This provides reproducible operation during routine manufacturing and becomes the foundation of process validation. The actual process control subset area within a design space is referred to as the control space, which is where a manufacturer would normally operate. Operating a process outside its control space but within its design space should be investigated but is not reported to the FDA (which is currently necessary for validated processes).
Regulatory Flexibility: Under the new operating paradigm, a design space is proposed by each manufacturer and is subject to regulatory assessment and approval. Process improvements (as part of continual improvement efforts) within the approved design space do not constitute a process change and thus can be implemented without further regulatory review. This degree of “operational freedom” within what is now being referred to as regulatory flexibility is predicated on an acceptable level of relevant scientific knowledge provided by a manufacturer in its registration application. Manufacturers thus envision a risk-based regulatory flexibility such that movement within each approved design space does not require traditional postapproval submittals (9, 10). Currently, the FDA and industry together are exploring a product-specific regulatory agreement as a venue to capture this much-expected flexibility (11).
Design Spaces for Unit Processing Operations: In general, biopharma–ceutical manufacturing processes involve four principle operations (see “Process Characterization” in the online appendices): cell culture, clarification, purification, and fill-finish, each harboring a number of distinct individual manufacturing steps referred to as unit processing operations (UPOs). Many of those process steps are governed by volume and time, among other factors.
The elucidation of relevant quality attributes and process parameters is highly specific to the nature of each UPO, so determining those that are relevant for establishing a design space is equally specific. Therefore, Table 1 merely provides a nonexhaustive list of example quality attributes for UPOs commonly used in biopharmaceutical manufacturing with operating and process parameters that can be evaluated for criticality in establishing design spaces.
Table 1: Example product quality attributes with operating parameters (e.g., equipment, materials, and procedures) and process parameters that can be evaluated for criticality in establishing design spaces for two common unit processing operations in biopharmaceutical manufacturing
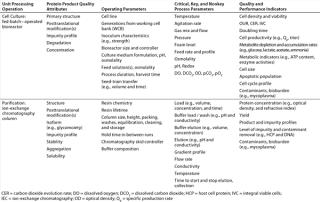
Following the principles of QbD, product and process specifications are directly related to CQAs, which are in turn linked to CPPs. Each design space with its embedded control space provides a scientific rationale to justify proposed acceptance criteria and truly represents the cornerstone of the new operating paradigm.
Process Understanding: Key to SuccessAn optimal process-design approach focuses on establishing process capability early in development of a design space as part of an overall strategy to arrive at a manufacturing process that is actually effective and robust from day one. It is based on knowledge gained from development studies as well as experiences gained from technology transfer and manufacturing.
For example, during development CQAs and CPPs are identified based on systematic experimental design (e.g., DOE) and often corroborated with prior knowledge, if available. It is the degree to which these critical attributes and parameters can be causally linked that ultimately determines the level of process understanding achieved. This subsequent escalation from data captured to valuable information and ultimately to product knowledge and process understanding is a pivotal outcome of any well conducted development program. This is a regulatory expectation.
Figure 3 defines five levels of process understanding ranging from the lowest (the descriptive level merely capable of describing what the process at hand is) to the highest “first principles” level combining experimental data and mechanistic knowledge based on chemistry, biology, physics, and engineering to model and predict performance. Although the highest level of process understanding is desirable, it is not required or even currently achievable in many cases. The “level two” process characterization represents a minimum r
egulatory-acceptable level.
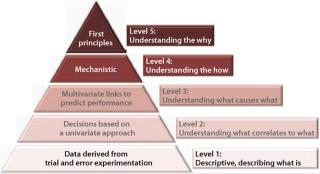
Because a bioprocess in essence defines its product, and aspects of the heterogeneity of each biomolecule defines its quality, a quantitative point-to-point distribution of product attributes and process parameters across a design space is needed to achieve complete product knowledge and process understanding (see “Product Characterization” in the online appendices). So it’s easy to understand why so much effort is currently being devoted to product characterization and process controls in biopharmaceutical manufacturing processes.
Quality Risk Management: The Missing LinkToday a great variety of specific QRM methods and tools exist in addition to basic risk-management facilitation tools (e.g., flow charts, process mapping, check sheets, and cause-and-effect diagrams) and supporting statistical methods (e.g., control and Pareto charts). Those formal QRM tools provide the opportunity for a common understanding and language of risk assessment in terms of probability (referring to the past), detectability (referring to the current), and severity (referring to the future) of harm through the use of structured process thinking.
Risk is defined here as a function of the probability of the occurrence of harm and its severity. It needs to be differentiated from uncertainty, which is best described as a lack of (or inadequate) knowledge. A specific risk question and its specific area of application (system, process, or product) will often provide insight regarding which tools to apply. Along with the level of detail sought and inherent organizational experience and preferences, this further contributes to tool selection. No single tool is appropriate for all cases.
Table 2 lists various formal QRM tools largely from the ICH Q9 (4) QRM briefing pack (12) that may be applied to different aspects of pharmaceutical quality by industry and regulators alike. Industry applications include product and process development, manufacturing, and distribution. Regulatory applications include inspection and submission/review processes throughout the life-cycle of drug substances and drug products.
Table 2: Nonexhaustive overview of formal quality risk management (QRM) tools useful in process design and manufacturing.

For example, use of formal risk-assessment tools in industry is valuable during initial development for identifying CPPs and raw-material attributes in developing control and risk mitigation strategies to minimize undesirable variation in CQAs. Subsequently, they are also powerful for predicting the effects of contemplated process changes on CQAs. Another application involves the use of formal QRM tools in assessing the need for additional studies (e.g., bioequivalence, stability) relating to scale-up and technology transfer. In manufacturing, they are helpful for evaluating the frequency and extent of in-process control testing and even in evaluating and justifying the use of PAT toward the goal of parametric and real-time release.
Risk control is an essential element of QRM, and the ability to formulate effective risk-control strategies is directly proportionate to the level of process understanding achieved. So the combination of QRM tools and process understanding guide process design and control activities. Together they help companies determine their degree of opportunity to prevent or mitigate the risk of producing poor-quality products as well as gauge the effectiveness of implemented quality systems and the degree of process robustness (see the online appendix). The effectiveness of risk-control strategies may be challenged, especially if compounded effects of multiple “risk” factors are poorly understood. Implementation of risk-control measures may in and of itself introduce new risks. For example, the installation of in-process measurement technology to alleviate or reduce previously identified risks can itself establish a new demand for risk management.
On the regulatory side, the FDA is progressively implementing risk-based approaches to modernize its review, compliance, and inspection components of pharmaceutical manufacturing and product quality regulations (13).
Overall, QRM efforts for industry and regulators alike should be guided by two principles:
-
Evaluation of the quality risks should be based on scientific knowledge and ultimately link to protection of patients
-
The level of effort, formality, and documentation of a QRM process should be in every application commensurate with its level of risk.
By helping companies establish well-understood and robust processes, QRM approaches could reduce overall regulatory burden through, for example, downgrading or eliminating certain types of postapproval supplements. This may be achieved in proportion to each manufacturer’s overall efforts to reduce and mitigate product quality risk (e.g., by managing sources of variability) and adhering to continual improvement.
Continual Improvement: Closing the LoopContinual improvement is an essential element of any modern quality system that aims at improving overall process quality and efficiency. This operates primarily by directing efforts toward reducing variation in process and product quality characteristics, yet it is not aimed at changing the fundamental design of a manufacturing process. In fact, when it is properly applied, a product and a process should already be in compliance with specifications, so improvement activities are directed toward incremental process optimization. The actual steps (such as adjustment of process parameters or operating schemes using more advanced control options) should be implemented within the original design space set forth by the original process. That is a critical consideration (14) because, regardless of the intent of a change, product quality and performance remain ensured only within the original design space.
The term continuous improvement is often used more broadly to encompass all improvement efforts, including correctiv
e actions and ensuing preventive actions (CAPA). From a regulatory perspective, however, a distinction between CAPA and other continuous improvement activities should be made because CAPA is needed only when product quality characteristics are out-of-specification (OOS) (15) or otherwise in question.
Both continuous and continual improvement must also be differentiated from innovation. Innovation applies to the application of fundamentally new approaches to process design and control through, for example, the use of entirely novel technologies such as those enabled by PAT — as compared with incremental modifications performed solely in routine production operations.