 Quality risk management (QRM) is a systematic process for assessment, control, communication, and review of risks to the quality of a pharmaceutical product across its lifecycle. Although QRM is not new (1), the regulatory focus on QRM will increase with the arrival of the European Medicines Agency’s (EMA’s) Annex 1 (2), which was reviewed by the US Food and Drug Administration (FDA), the World Health Organization (WHO), and the Pharmaceutical Inspection Convention Scheme (PIC/S). Integrity testing of sterilizing-grade filters is a key focus of QRM because it is a fundamental element of sterility assurance.
Quality risk management (QRM) is a systematic process for assessment, control, communication, and review of risks to the quality of a pharmaceutical product across its lifecycle. Although QRM is not new (1), the regulatory focus on QRM will increase with the arrival of the European Medicines Agency’s (EMA’s) Annex 1 (2), which was reviewed by the US Food and Drug Administration (FDA), the World Health Organization (WHO), and the Pharmaceutical Inspection Convention Scheme (PIC/S). Integrity testing of sterilizing-grade filters is a key focus of QRM because it is a fundamental element of sterility assurance.
The following discussion points out the insufficiency of traditional QRM for filter-integrity testing and advocates for a comprehensive approach. Such a method offers thorough identification of possible failure modes, ways to prevent failure, and improved detectability through use of program-specific parameters for automatic detection of abnormal conditions.
Why QRM?
More than just a regulatory requirement, QRM must be implemented for patient safety. Filter-integrity testing is an essential step for lot release. A false-passed integrity test (e.g., a conforming test result even though a filter is broken) could jeopardize patient health if it is not detected through required sterility testing. A false-failed integrity test (a failing test result despite filter integrity) would require drug quarantine, incurring a negative financial impact for the manufacturer. Worse, it could jeopardize patient health by interrupting supplies of an essential medication.
Traditional QRM
Everything that mitigates risk for false-passed and false-failed test results can be considered as part of QRM and must be maintained. Training issues constitute one key element. Operators must be sensitive to the importance of reliable filter-integrity testing and to the impact of false test results on both a manufacturer and patients. Operators must be trained to conduct testing under prerequisite conditions and to detect abnormal test results. Quality assurance (QA) personnel also must know how to detect abnormal test results. Using a barcode scanner to select the correct test program minimizes the risk of running the wrong test, and personnel must follow standard operating procedures (SOPs).
However, end users often report that those measures are insufficient and not enforced well enough to prevent serious deviations. Among the problems encountered is that a program can be defined with the wrong parameters. Examples include incorrect diffusion, forward flow, or intrusion test pressure; inadequate stabilization time; and an incorrect minimum bubble point (BP). In some cases the entirely wrong program is applied, including use of the wrong filter type or size. A faulty connection to the filter setup can compromise test results. For example, if a downstream valve is closed, a filter will not be tested at the correct differential pressure. Some users report problems related to testing filters in the reverse direction, environmental temperatures lying outside the defined range, the temperature of a wetting liquid falling out of range, and temperature fluctuations in general. Finally, the wrong test gas (e.g., nitrogen instead of air) can be used.
Such failure modes can generate both false-passed and false-failed test results, which must be prevented to keep nonsterile drugs from reaching the market and to obviate drug quarantines. Another reported issue is that some users conduct irregular numbers of test repeats before reaching a passed test result — indicating that a test procedure is not under expert control and/or that the operators are not sensitized to the consequences of quality deviations. In fact, regulatory bodies might be concerned that an irregular number of test repeats indicates a lack of data integrity — and perhaps even fraud.
Automated QRM
Recent developments enable significant improvements in prevention and detectability of failure modes in filter-integrity testing. Identifying normal test values for each filter setup and defining program-specific parameters can help systems detect abnormal conditions and faulty setups automatically. Examples of such parameters are determined below.
Expected Minimum and Maximum System Net Volume: During programming of the Sartocheck 5 Plus filter tester, it’s possible to enter minimum and maximum expected values for the system net volume of a filter setup. If the tester detects a volume outside that defined range, the test is interrupted automatically.
An upstream net volume could fall outside the specified range for a number of reasons:
- The wrong filter size is being tested
- The right filter is being tested, but in the wrong direction (volume is therefore too small)
- A valve is closed between the integrity tester and the filter, which also results in too small a volume
- A broken filter and closed downstream valve result in too large a volume
- The setup is incorrect (e.g., an operator forgets to install a protective hydrophobic filter between the integrity tester and the filter to be tested).
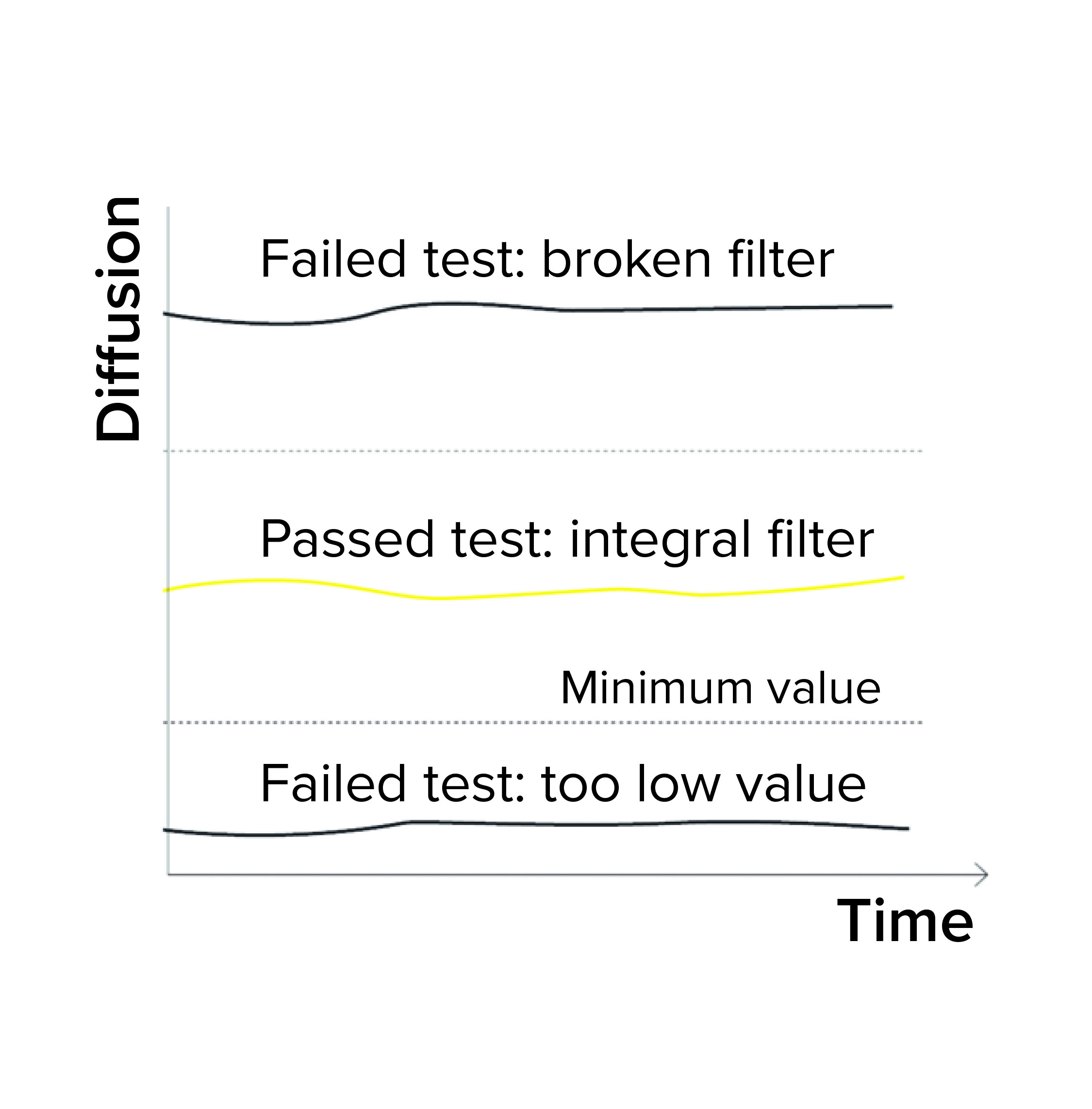
Figure 1a: Diffusion/water intrusion test
Expected Minimum Diffusion: When programming a diffusion test or a combined diffusion and BP test, a user can enter a minimum expected diffusion value per filter/sample in addition to a validated maximum diffusion value. If the integrity testing device measures a value below the minimum diffusion, the test fails (Figure 1a). Diffusion could fall below an expected minimum value because of the following conditions:
- Too small a filter is being tested
- A valve is closed between the integrity tester and the filter to be tested (which would be detected by the volume measurement)
- The wrong type of filter is being tested even though it is the correct size (e.g., using a double-layer instead of a single-layer membrane)
- A valve on the downstream side is closed, so pressure is building on that side
- The membrane is abnormally clogged
- The filter is being tested with the wrong temperature or type of wetting liquid.
Expected Minimum Water Flow Value: When programming a water-intrusion test, a user can enter a minimum expected water flow value for each filter/sample. If the integrity testing device measures a value below that minimum intrusion, the test is reported as failed (Figure 1a). Reasons for the intrusion test to fall below a minimum value include the following:
- Too small a filter size is being tested
- A valve is closed between the integrity tester and the filter to be tested (which would be detected by the volume measurement)
- The wrong type of filter is being tested even though it’s the correct size (e.g., a double-layer instead of a single-layer membrane)
- The filter (although of a correct type) is broken, with a closed downstream valve and no water flow
- The filter is being tested with water that is too cold.
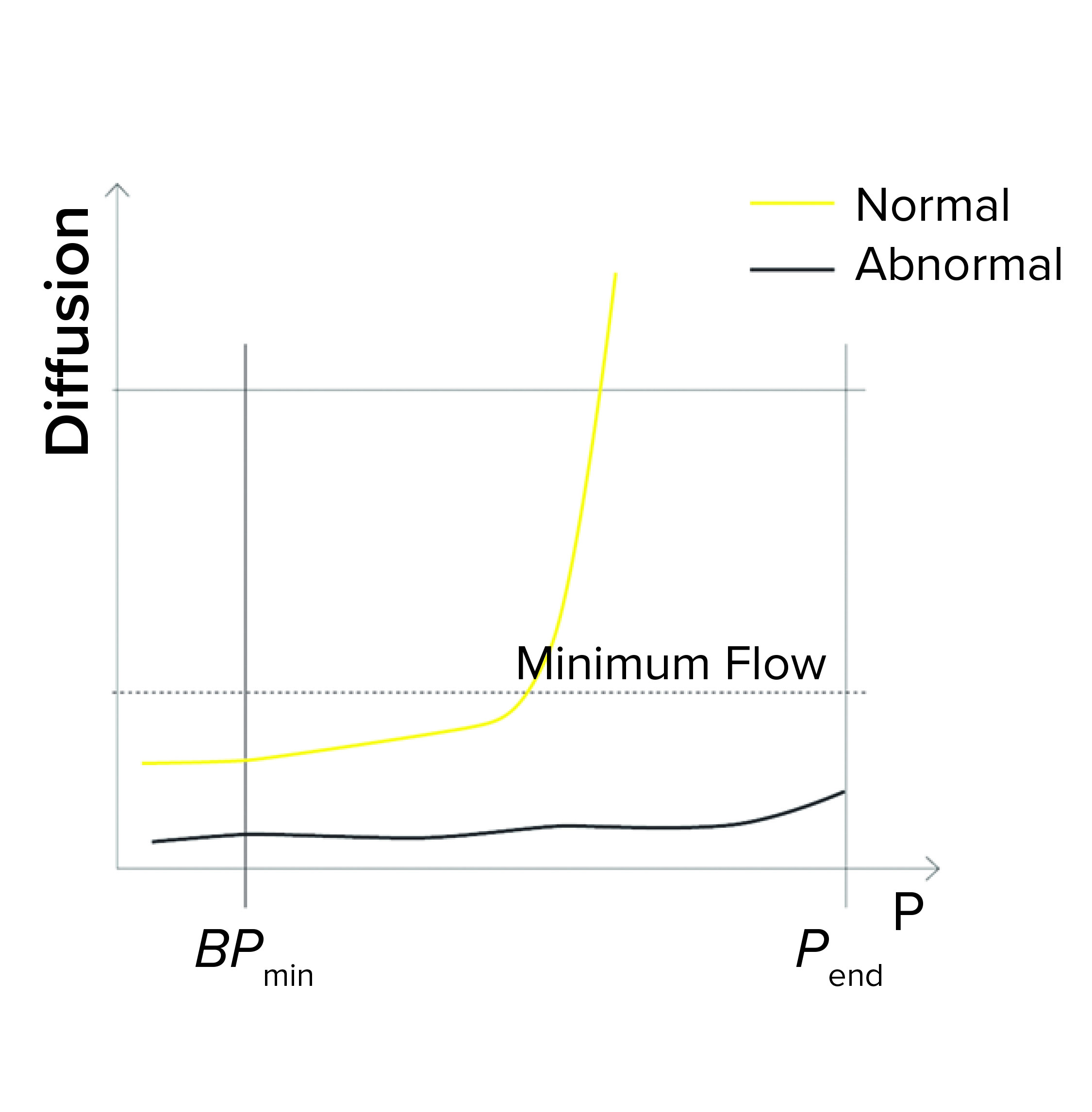
Figure 1b: Bubble point (BP) test
Minimum Flow at the End of a BP Test: During programming of a BP test or a combined diffusion and BP test, a user might enter a minimum expected flow at the end of the test — so that the end pressure (Pend) is defined during programming. If that measured flow is below the minimum when Pend is reached, the test reports as failed (Figure 1b). Reasons for the flow to fall below the limit at the end of the test include the following:
- The wrong filter type is being tested (e.g., it has too small a pore size, and thus a higher BP value)
- A closed downstream valve causes pressure to build on the downstream side
- The membrane is abnormally clogged
- The filter is being tested with the wrong type of wetting liquid.
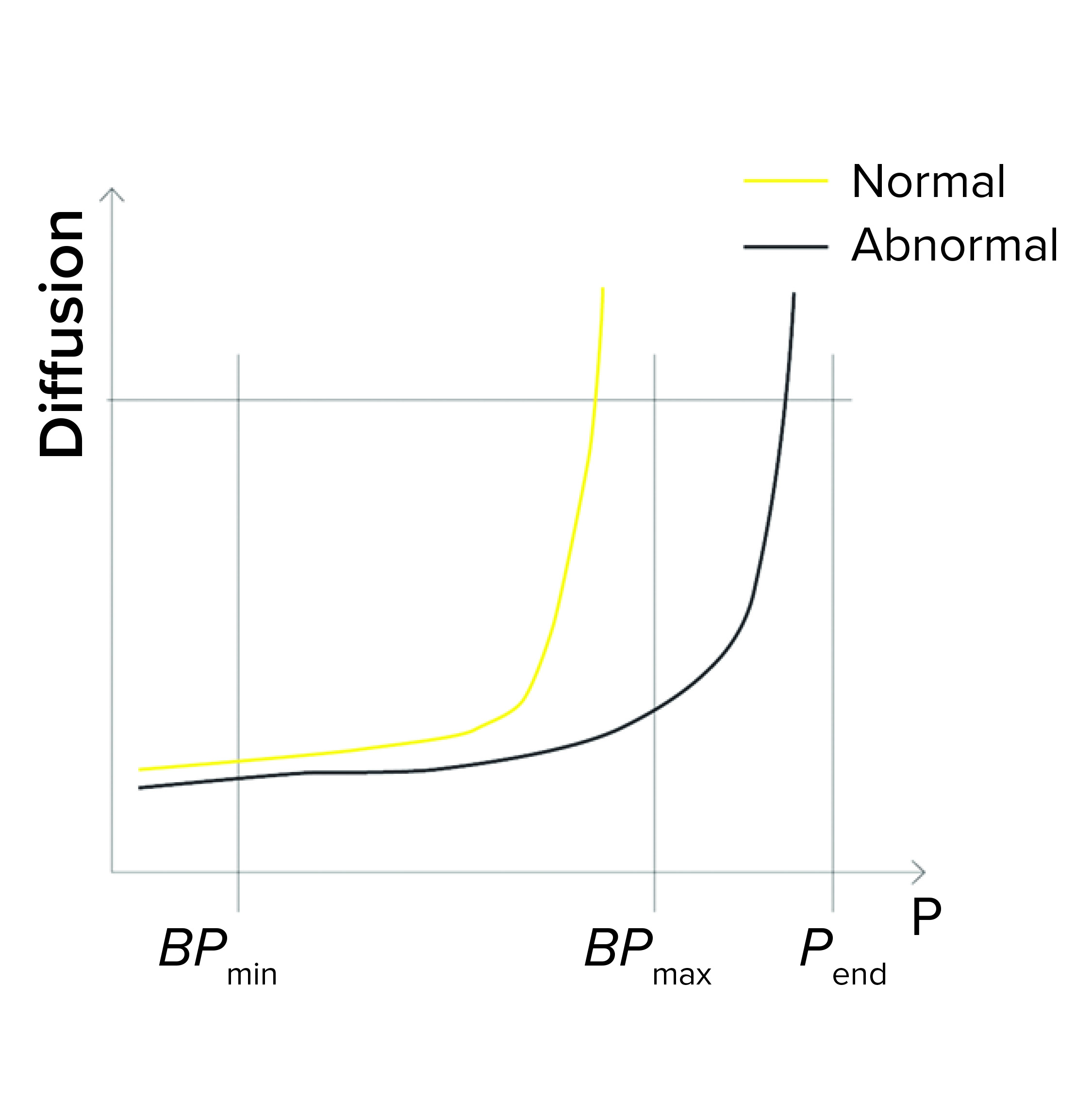
Figure 1c: Bubble point (BP) test
BPmax Is Set to Lower Than Pend: During programming of a BP test or a combined diffusion and BP test, a user can set the BPmax value to a lower value than pressure at the end of the test (Pend) so as to detect abnormally high BP values (Figure 1c).
If the actual BP is detected above BPmax (abnormally high), the test fails. That can happen because
- The wrong filter type is being tested (e.g., it has too small a pore size and thus a higher BP value)
- A closed downstream valve is causing pressure to build on the downstream side
- The membrane is abnormally clogged
- The filter is being tested with the wrong temperature or type of wetting liquid.
Additional Generic Automated QRM
Some failure modes are independent of the filter setup and thus are generic rather than program-specific.
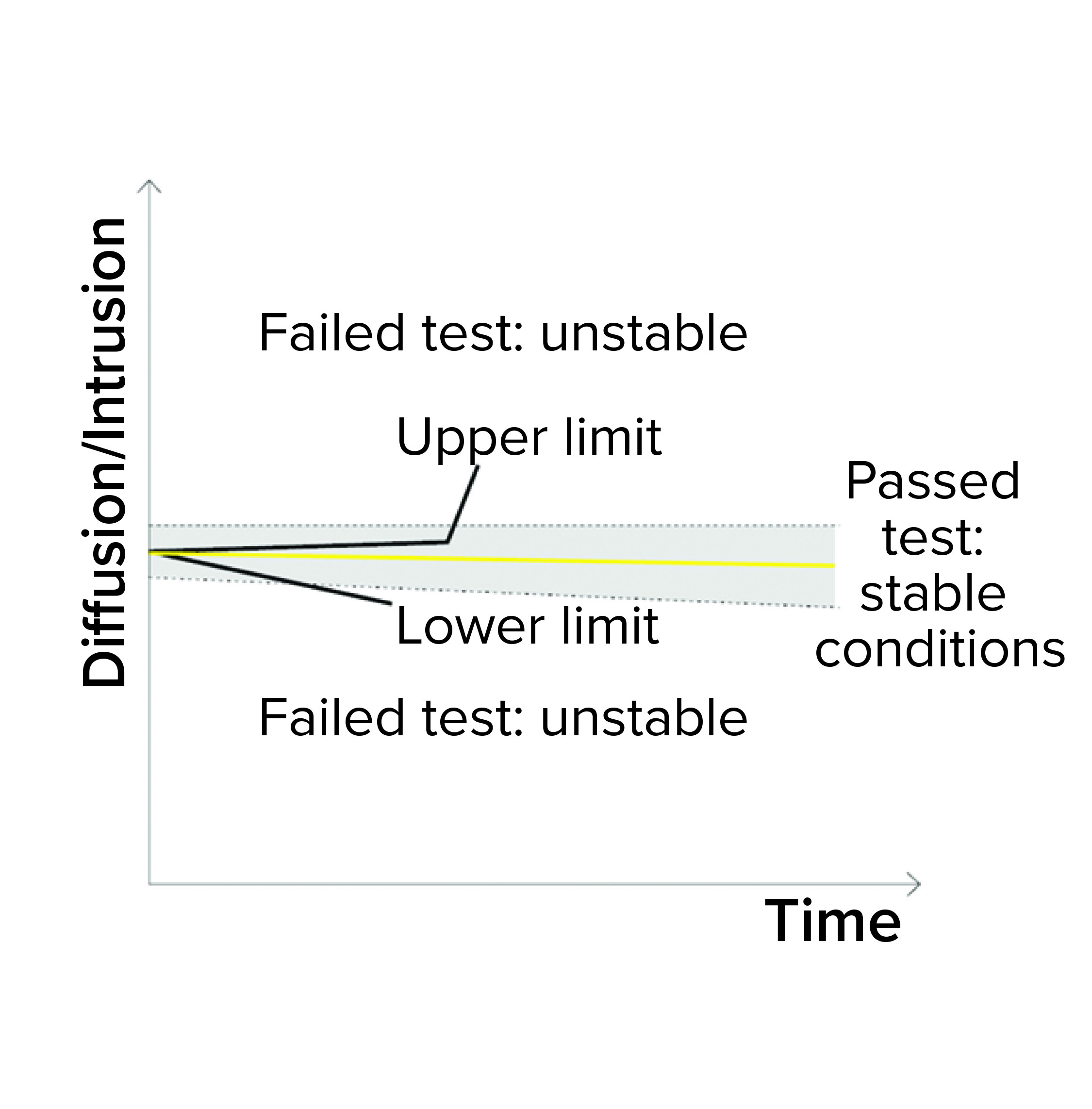
Figure 1d: Bubble point (BP) test
Detection of Unstable Test Values: A filter system should have a stable value during the diffusion/water intrusion measurement if it has been prepared with a wetting liquid at the appropriate temperature, without any closed valve on the downstream side, and with an appropriate stabilization time (Figure 1d). In the case of a BP measurement, the diffusion values should increase slightly as the pressure increases. Any deviation could be attributed to a failure mode, including under the following conditions (Figure 1e):
- The filter is being tested with wetting liquid that is too cold or too warm, generating temperature changes inside its housing
- There are environmental temperature variations
- A downstream valve is closed.
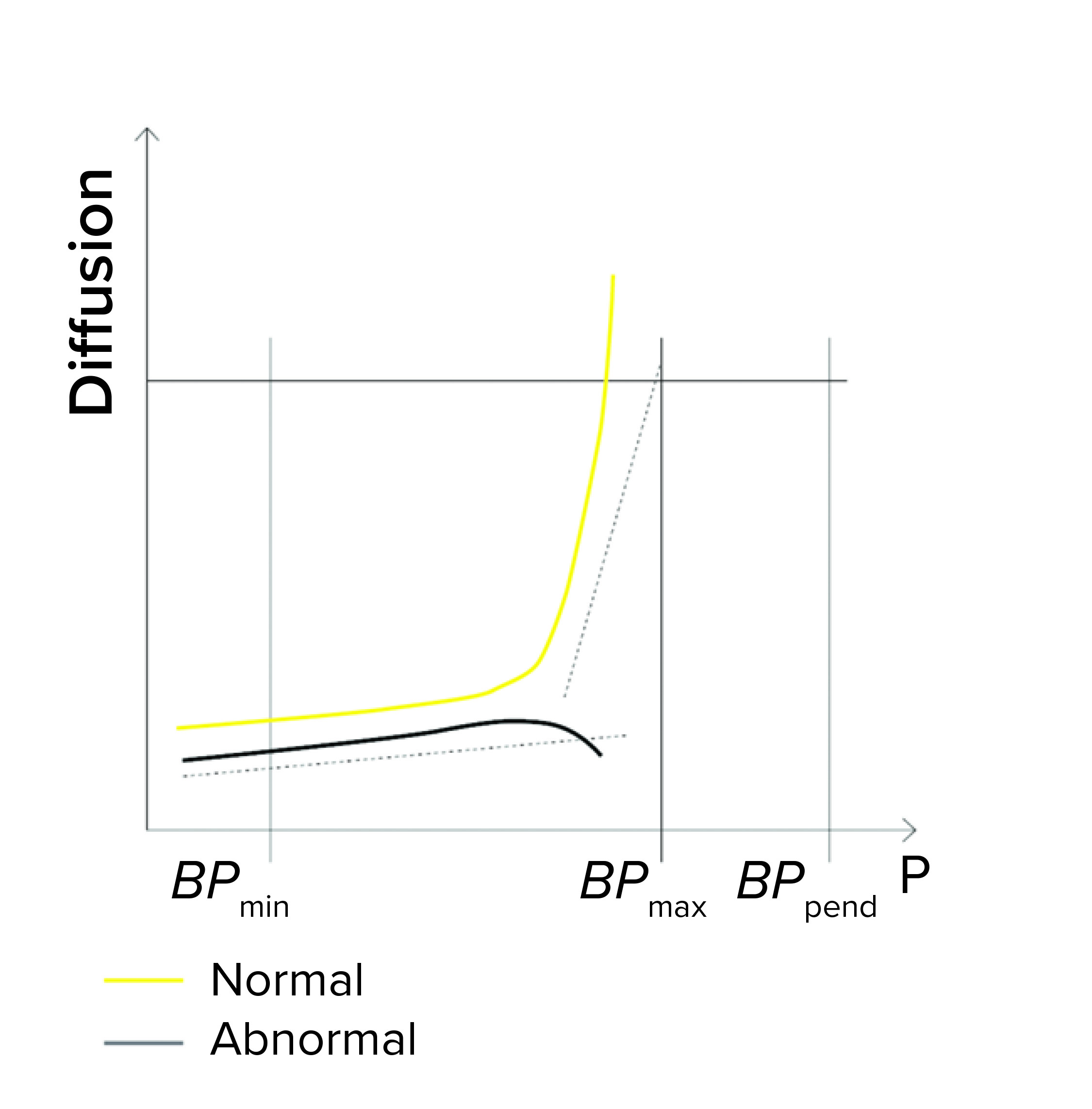
Figure 1e: Diffusion/water intrusion test
Detection of an Environmental Temperature Outside Specifications: An environmental temperature that falls outside of specification is a major concern for reliable integrity testing (3, 4). Also, an integrity test could be performed at a stable temperature but one that is too warm or cold.
Other temperature fluctuations can be caused by poor ventilation by the laboratory’s heating, ventilation, and air conditioning (HVAC) system; by effects from steam-sterilizing surrounding equipment; and by exposure to direct sunlight. Placing a temperature sensor right next to a filter setup being tested and connected to the integrity tester can prevent starting a test if an environmental temperature is out of specification. Such a practice greatly improves detectability of environmental temperature variations that otherwise could generate false passed/failed test results.
Stainless-steel (traditional housings) and plastic (single-use capsules made of polypropylene) filter setups will react differently to temperature variations (4). Adapting risk assessment related to temperature variations based on both the type of filter setup and exactly when a variation occurs reduces the risk of both false-pass and false-fail results.
Preventing Irregular Test Repeats
The Parenteral Drug Association (PDA) recommends that a filter should be tested no more than three times at an end-user site (5, 6). Regulatory inspectors tend to view registering more than three tests as evidence of a process that is not under control. Note that executing more than three tests but registering only three of those can lead regulators to question data integrity and perhaps to suspect fraud. It is essential that operators understand such repercussions.
Relying on audit-trail verification is insufficient justification because audit trails can contain massive amounts of events and information. By the time a quality deviation is discovered, it already will have become a fact. The safest approach is to prevent operators from executing more than the recommended number of test repeats through intelligent test counting and actually blocking additional tests.
Intelligent test counting also is necessary for excluding irrelevant tests (e.g., those that have been aborted because of faulty setups). This feature will be implemented through a Sartocheck 5 Plus software release in 2021. The software will come with a comprehensive risk assessment documentation allowing for a secure upgrade of existing Sartocheck 5 Plus devices.
Preventing Cross-Contamination
A filter-integrity test device should last for several years. From the first day of use to the last, it must not release uncontrolled bioburden or particles into filters being tested.
Filters that are tested postuse (and in some cases, preuse) can be tested with an actual product as the wetting liquid. Even if rinsed with water, in the case of a postuse test, a filter can contain traces of product. Any liquid siphoning by the integrity tester or aerosols flowing back into the test tube or the device pneumatics are likely to be pushed back during subsequent integrity tests. If a contaminated integrity testing device is used for preuse testing, the tested filter is likely to become contaminated, severely compromising quality.
To mitigate cross-contamination, the Sartocheck 5 Plus filter tester can be used with an accessory kit for external venting, thus preventing siphoning of aerosols or liquids. One or several such kits can be assigned to a given tester, and each program can be defined to be used with or without the accessory kit. In addition, a cleaning kit soon will be available for separate cleaning of the accessory kits and internal tester pneumatics with up to 0.5 M NaOH. Another way to mitigate cross contamination is to have dedicated integrity test devices for each application. That practice would not reduce the benefit of periodical cleaning of the internal pneumatics.
QRM Handbook: Conducting FMEA
Failure modes and effects analysis (FMEA) is used commonly for QRM whenever simple quantification of risk would be insufficient and when identification of root causes and means of mitigation are paramount.
FMEA is one of the most effective tools for mitigating risk. The aim of an FMEA for filter-integrity testing should be to identify operator hazards, risks for false-passed and-failed test results, and threats to the functionality of a device as closely as possible to their source. FMEA should give extensive information on how to improve filter-integrity testing from a general perspective, how to optimize the use of a device, and how to approach detection of faulty setups to prevent false-passed test results (7).
A QRM handbook for the Sartocheck 5 Plus test device provides guidance for setting up a comprehensive FMEA for filter-integrity testing and also contributes to improved operator safety (HSE/OSH). It is available on request.
Impact of an Unlikely Calibration Offset
QRM requires all events that could have an impact on quality to be evaluated and mitigated. Accuracy of the pressure sensor in a filter-integrity device is an essential aspect of quality assurance in lot release (8). If the annual calibration reveals a pressure reading offset outside of specifications, then a common conclusion is to question the accuracy of all integrity test results from the past year. Even if a calibration offset in the pressure sensor of a high-quality integrity tester is unlikely, the severity of such an event is so high that QA divisions need an appropriate tool to estimate the impact of a calibration offset on integrity-test results. The impact of a calibration offset on the accuracy of algorithms used by the Sartocheck 5 Plus tester has been documented (8), and a tool has been developed to quantify this impact.
 Improving Quality Risk Management
Improving Quality Risk Management
The arrival of the Annex 1 emphasizes that processes, equipment, facilities, and manufacturing activities for all sterile pharmaceutical products and sterile active substances should be managed in accordance with QRM principles. QRM also applies to filter-integrity testing because it is an essential element of sterility assurance. As revealed though evaluating customer complaints, drug developers cannot rely only on traditional QRM approaches such as operator training, bar-code scanners for program selection, and SOPs.
Program-specific safety parameters on the Sartocheck 5 Plus integrity tester have made it possible to prevent certain failure modes and thus greatly have improved their detectability to protect patients in compliance with Annex 1. Comprehensive FMEA documentation also should be viewed as a contributor to improving QRM in drug manufacturing.
References
1 ICH Q9. Quality Risk Management. International Council for the Harmonisation of Technical Requirements for Pharmaceuticals for Human Use: Geneva, Switzerland, 2005; https://database.ich.org/sites/default/files/Q9%20Guideline.pdf.
2 EU Annex 1, Consultation Document: Manufacture of Sterile Medicinal Products. European Commission: Brussels, Belgium, 2020; https://ec.europa.eu/health/sites/health/files/files/gmp/2020_annex1ps_sterile_medicinal_products_en.pdf.
3 Stering M. Failure Mode Effects Analysis for Filter Integrity Testing. Pharma. Technol. 40(4) 2016: 66–69; https://www.pharmtech.com/view/failure-mode-effects-analysis-filter-integrity-testing-0.
4 Stering MA. Risk Assessment for Thermal Influences on Filter and Container Closure Integrity Testing. Pharma. Eng. 37(4) 2017: 56–61; https://ispe.org/pharmaceutical-engineering/july-august-2017/risk-assessment-thermal-influences-filter-container.
5 Antonsen HR, et al. Technical Report 26: Sterilizing Filtration of Fluids. PDA J. Pharm. Sci. Technol. 62(5 suppl.) 2008: 2–60.
6 Points to Consider for Aseptic Processing: Part 2. Parenteral Drug Association: Bethesda, MD, 2016; https://store.pda.org/TableOfContents/43527_TOC.pdf.
7 Sartocheck® 5 Plus Filter Tester. Sartorius AG: Göttingen, Germany, 2020; https://www.sartorius.com/en/products/process-filtration/hardware/integrity-testing/sartocheck-5.
8 Stering M. Effects of Pressure Sensor Calibration Offset on Filter Integrity Test Values. BioProcess Int. 11(10) 2013: 46–53; http://lne.e92.mwp.accessdomain.com/downstream-processing/filtration/effects-of-pressure-sensor-calibration-offset-on-filter-integrity-test-values-347994.
Magnus Stering is senior product manager, Integrity Testing Solutions, at Sartorius: magnus.stering@sartorius-stedim.com, www.sartorius.com.