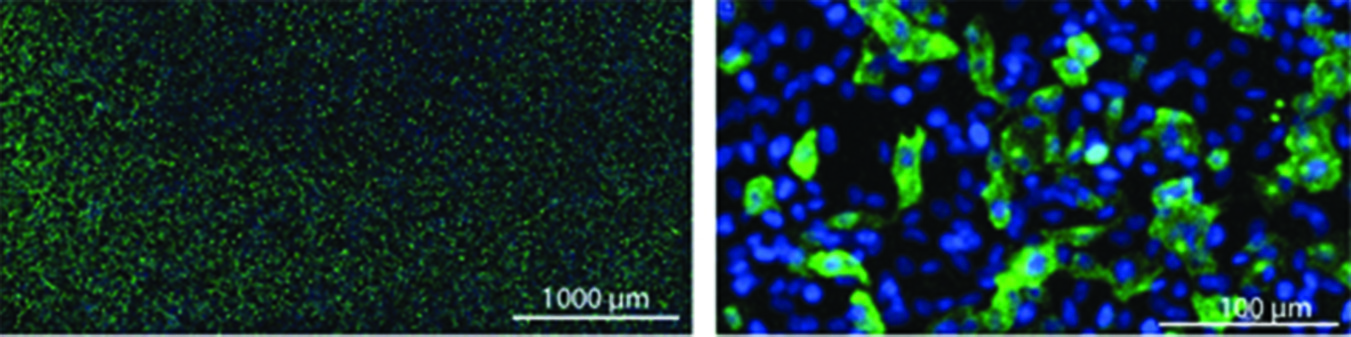
Vero cells infected with purified influenza A: Infectious virus particles were stained with rabbit antiinfluenza A NP-antibody and a goat antirabbit IgG antibody conjugated with AlexaFluor 488 (green). Cell nuclei are stained blue with DAPI (4‘,6-diamidino-2-phenylindole).
Influenza is a global respiratory disease with an estimated mortality of up to a half million people per year (1). The majority of traditional influenza vaccines are still produced in eggs. Downstream processing typically consists of clarification by centrifugation, concentration by ultrafiltration, and purification by ultracentrifugation (2).
Recombinant vaccines are most often purified by chromatography. Chromatographic purification of viruses already has achieved major improvements in recovery and scalability (3), but it also is important because it enables virus purification to keep pace with important regulatory and manufacturing trends across the field of biopharmaceuticals. One of those trends is process intensification, referring to development of processes that harmonize integration of fewer and more capable steps to achieve higher productivity and reproducibility as well as reduce manufacturing costs.
Intensification takes many forms. Continuous processing represents one aspect, and it already has been applied successfully to anion-exchange purification of influenza A (4). An arguably more important aspect involves application of tools that overcome limitations of traditional options and thereby improve results and/or reduce the number of process steps. Application of single-use processing materials also contributes to intensification. By suspending the validation and manufacturing burdens of multiple use components, disposables help companies focus resources on essential process functions.
As with many viruses, purification of influenza virus from clarified cell culture harvests often begins with tangential-flow filtration (TFF), which simultaneously achieves multiple tasks. The first is concentration of the virus, which is retained, while contaminant levels are reduced by passing through membrane pores. Both capabilities often are referred to as ultrafiltration (UF). The third capability is buffer exchange by a process called diafiltration (DF). The original fluid is replaced gradually by buffer with a formulation suitable for an initial purification step. In this discussion, reference to UF–DF is simplified to TFF.

Figure 1: Shear stress in the void volume of packed particle columns; solid areas represent particle surfaces. Interparticle void space is indicated in white. Black arrows indicate flow, and line thickness indicates flow velocity. Circular patterns indicate eddies. Areas marked in red highlight zones of countercurrent flow that generate shear stress. Axial shear stress at particle surfaces is negligible with flow rate approaching zero there because of friction.
Despite the valuable role of TFF, its benefits come at a price. Shear stress produced during TFF has been documented to strip outer envelopes from lipid-enveloped viruses and to fracture brittle capsids of non-lipid−enveloped species (5, 6). Such liabilities might be prevented by direct virus capture on traditional porous particle columns, but this is impractical for two reasons. An obvious reason is the extended time that would be required to load unconcentrated virus. The other is that chromatography columns packed with particles represent an additional source of shear stress. Flow through the irregular spaces between particles causes formation of eddies, zones of recirculation. When bulk flow through the interparticle space encounters eddy flow in the opposite direction, it generates shear stress at their interface (Figure 1). Shear stress in column void spaces is directly proportional to flow rate (7).
One key distinction of monolithic chromatography media compared with columns packed with particles is that monoliths do not have void spaces. Flow through the interconnected channels is laminar, so eddy formation and shear stress are nil. Another key distinction is that mass transport in monoliths is exclusively convective, rather than the situation in porous particle columns where flow between particles is convective, but solutes move into and out of the pores exclusively by diffusion.
Diffusion is the reason columns packed with particles are slow. Diffusion is even slower for large products such as viruses because diffusion constants become slower with
increasing particle size. By contrast, convective flow can be thought of as a river. Objects flow at the same rate as the current no matter what size they are. The result is that binding efficiency, dynamic binding capacity, and elution efficiency in monoliths are all independent of product size and flow rate. Flow rates can be 20–50× higher than in packed-particle columns without compromising performance (8).
Altogether, the combination of convective efficiency and laminar flow enables rapid virus concentration on monoliths without the flow-rate restrictions of traditional columns and without the shear forces created by either TFF or particle-packed columns. In short, they enable the sequential functions provided by TFF and traditional column capture to be combined into a single faster process step with less risk of damaging a product.
In this report we describe processes for purification of influenza A and influenza B, both lacking TFF steps, and both using a single chromatography step with a cationexchange monolith on a single-use basis. The choice of process buffers enables final formulation by simple dilution of the product pool. DNA digestion requires two hours. Capture, purification, and formulation are achieved within four hours. Host-cell DNA and host-cell protein (HCP) are reduced more than 99%, and final virus recovery is 80%.
Materials and Methods
Influenza virus was produced in Vero cells. Viruses were constructed as previously described (9) and propagated in Nunc Cell Factory CF-10 systems (Thermo Fisher Scientific) using serum-free medium Opti Pro SFM (Invitrogen, Thermo Fisher Scientific) supplemented with 4 mM GlutaMax I (Invitrogen, Thermo Fisher Scientific). Upon infection, cells were grown using recombinant trypsin (Thermo Fisher Scientific) and incubated at 5.0% CO2 and 95% relative humidity at 37 °C (influenza A) or 33 °C (influenza B) for three days. Harvests were clarified by low-speed centrifugation. Clarified harvest was treated with 20 U/mL Benzonase endonuclease (Merck, Germany) in the presence of 2 mM magnesium for two hours at room temperature.
Virus purification was conducted with 1, 8, or 80 mL CIMmultus™ SO3 cation-exchange monoliths. They were sanitized in advance with 1 M NaOH, then washed with water and equilibrated to 50 mM HEPES, 200 mM sucrose, pH 7.0. Influenza A virus was diluted 2:1 (sample:buffer) and applied to the monoliths without further preparation. Influenza B virus was diluted 1:1 with equilibration buffer, then loaded at the same flow rate. The monolith was washed with 50 mM HEPES, 200 mM sucrose, pH 7.5 containing 100 mM NaCl (influenza A) or 50 mM NaCl (influenza B) and eluted with a linear gradient. The gradient endpoint buffer in both cases was 50 mM HEPES, 200 mM sucrose, 2.0 M NaCl, pH 7.5. Specific gradient configurations for influenza A and B are discussed in the next section. Columns were cleaned and sanitized in place with 1 M NaOH, 2 M NaCl after each run. All chromatography steps were performed at a flow rate of five bed volumes per minute. Infectious virus– containing fractions were diluted to the formulation salt concentration, and a nonanimal-derived stabilizer was added.
Infectious particle concentration was determined by focus-forming assay (FFA) using a Cytation 5 multimode fluorescent reader using influenza-specific antibodies. Briefly, FFA analysis was performed by dispensing fivefold sample dilutions on Vero cells growing in 96-well flat-bottom plates (Thermo Fisher Scientific). After incubation for 24 hours, cells were fixed and immunostained using an antibody specific for influenza virus NP. The number of infected cells was counted in triplicate for each sample. FFA titer was calculated and expressed as FFU/mL. DNA concentration was determined by using a Picogreen dsDNA assay kit or with qPCR. Total protein content was determined using a Coomassie (Bradford) protein assay kit (Thermo Fisher Scientific). Host-cell protein (HCP) content was analyzed by enzyme-linked immunosorbent assay (ELISA).
Results
Omitting TFF in advance of chromatography required that samples be equilibrated by dilution. Filtered harvest was diluted at a proportion of one part equilibration buffer to two parts sample, then applied to the equilibrated cation exchanger. This resulted in 0.4% of the virus particles in the flow through for influenza A and 3.5% for influenza B. Increasing the dilution to a proportion of 1:1 reduced influenza B losses in the flow through to 0.1% or less. With this approach, binding capacity of >3.16 × 1010 FFU/mL SO3 resin was reached, while achieving >99% reduction of host-cell DNA and HCP.

Figure 2: Elution of Influenza B from a 1-mL CIMmultus SO3 monolith — the inset represents a zoomed image of the elution step; blue = UV absorbance at 280 nm; red = UV absorbance at 260 nm; black = conductivity
Figure 2 illustrates the elution profile of influenza B from a 1-mL CIMmultus SO3 monolith with 1:1 dilution of the initial material and column equilibration at pH 7.0. The dominant feature is the extended load interval at 600 column volumes (CV). Because of the 5 CV/min flow rate, however, it corresponds to a loading time of only two hours. Following a brief wash step with pH 7.5 buffer containing 50 mM NaCl, the virus was eluted with a 50-CV linear gradient to 100% buffer B. The six-hour total chromatography time included sanitization, equilibration, load, wash elution, and cleaning in place (CIP). The infectious virus was recovered in a 4.1-mL fraction (indicated in the colored background area) representing an overall concentration factor of 73-fold from the undiluted cell culture. Virus concentration was 2.43 × 1010 FFU/mL, and recovery was 74%. The virus fraction was formulated with about a fourfold dilution to lower buffer capacity, adjust salt concentration, and introduce a nonanimal−derived stabilizer before final sterile filtration.
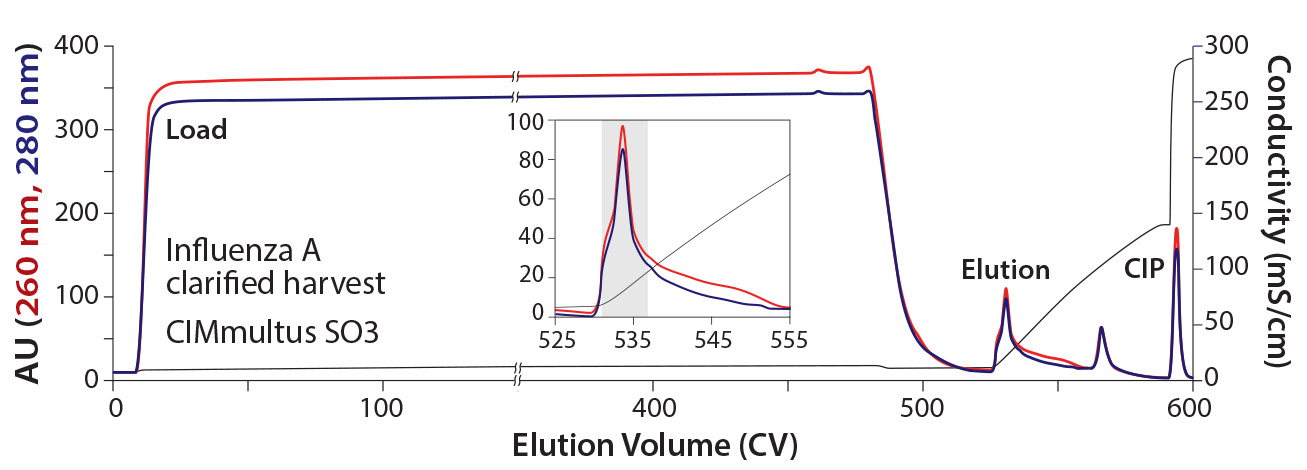
Figure 3: Elution of Influenza A from a 1 mL CIMmultus SO3 monolith — right image represents a zoomed view of the elution step; blue = UV absorbance at 280 nm; red = UV absorbance at 260 nm; black = conductivity
Figure 3 shows the elution profile of influenza A from a 1-mL CIMmultus SO3 monolith. Initial material was diluted 1:2 (buffer:sample) with equilibration buffer at pH 7.0. Similar to the influenza B process, loading volume was >500 CV. After loading, the column was washed using 100 mM NaCl containing HEPES buffer at pH 7.5. The product was eluted during 50-CV linear salt gradient to 2 M NaCl. Infectious virus was recovered in a 5.1-mL fraction (indicated in the colored background area) with concentration of 4.44 × 108 FFU/mL. Pooled infectious virus was formulated as described above. Overall recovery was 82%.
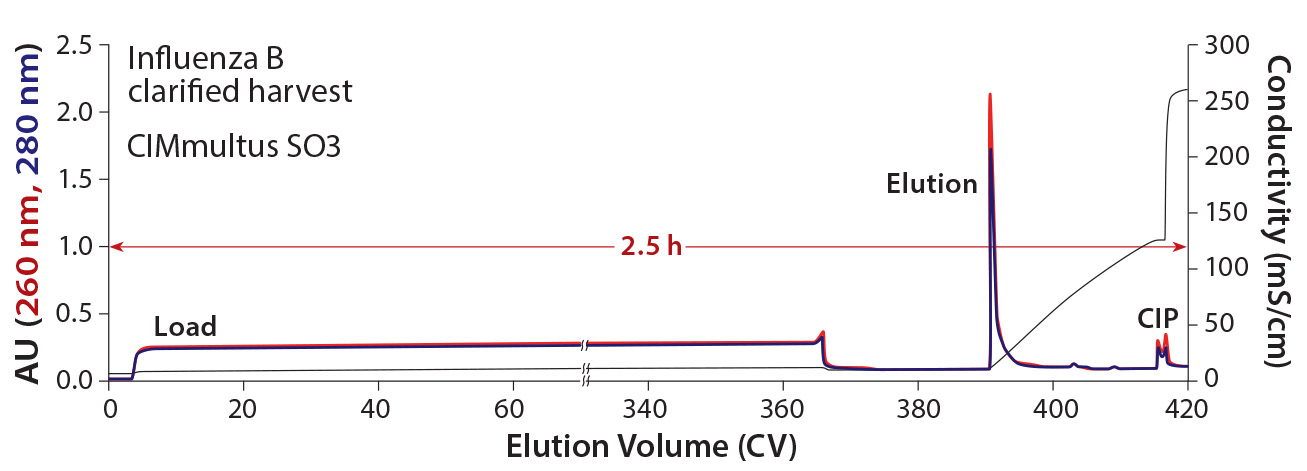
Figure 4: Elution of Influenza B from an 80 mL CIMmultus SO3 monolith with 2 µm channels; blue = UV absorbance at 280 nm; red = UV absorbance at 260 nm; black = conductivity; CV = column volume
The influenza B purification was scaled up 80-fold with an 80-mL CIMmultus SO3 monolith to supply material for clinical trials (Figure 4). About 360 CV of 1:1 diluted Benzonase-treated material was loaded at a flow rate of 3 CV/min, followed by a wash step with 50 mM HEPES, 50 mM NaCl, pH 7.5. Product was eluted during a 25-CV linear salt gradient to 2 M NaCl. Total chromatography time was four hours. Infectious virus recovery was 83.4%. Host-cell protein was reduced 99.7% and host DNA by 99.99%.
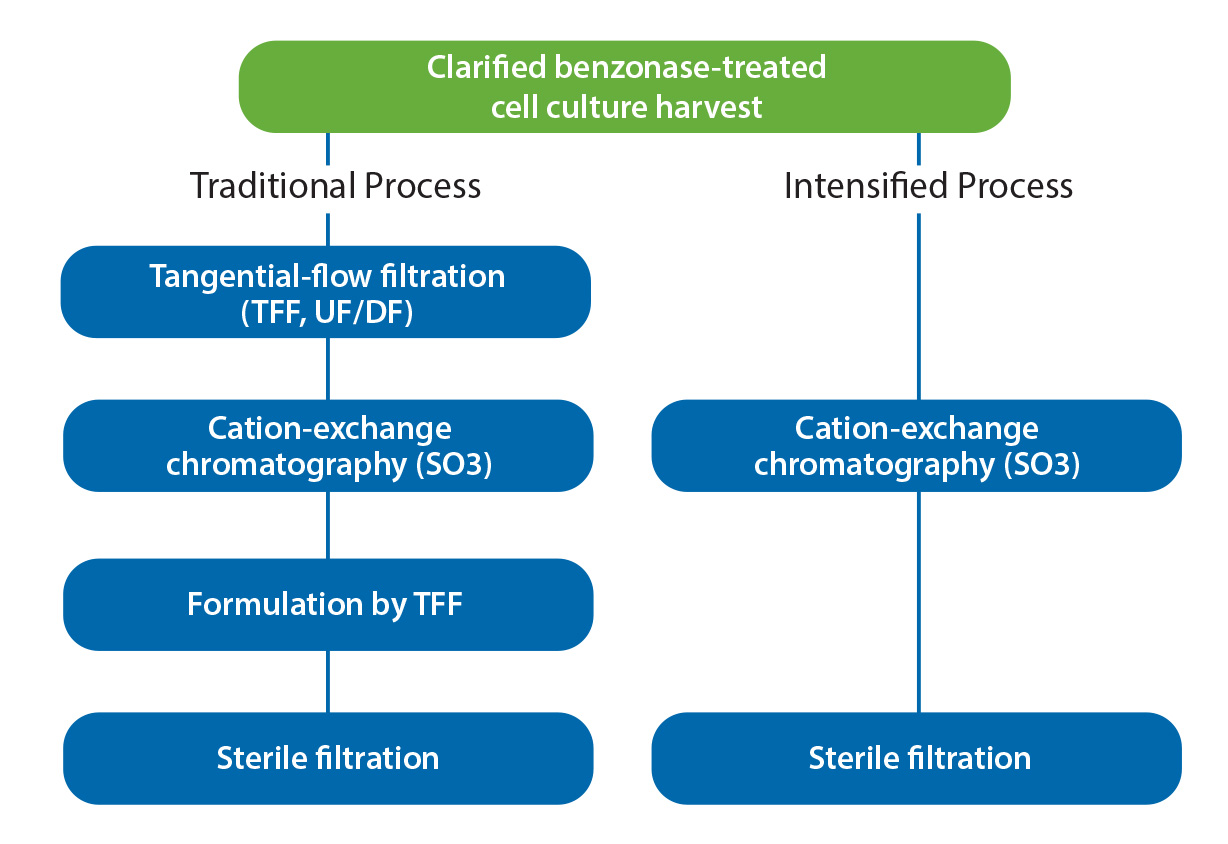
Figure 5: Instrument-operational steps in purification of influenza virus
Discussion and Conclusions
Direct virus capture from diluted feed streams with monolithic columns makes worthy contributions to process intensification during product development as well as during scale-up and manufacturing (Figure 5). Cell culture harvests for purification process development often are very limited in the early stages. Small-volume TFF systems are becoming more available but still not at a size to provide scalable results from laboratory scale to manufacturing. Use of a larger-than-necessary filter area increases risk of product loss because many virus species are known to bind nonspecifically to membrane surfaces (10). Neither of these liabilities is a factor with dilution-to-direct capture, and omission of membrane concentration bypasses a documented source of virus particle damage by shear stress.
Capture on a porous particle column still would require a TFF concentration step because of the low flow rates, but the ability of the monolith to accommodate high flow rates without compromising performance suspends that limitation. That ability to achieve concentration and purification simultaneously also bypasses two sources of shear stress and makes use of monoliths even more attractive. This is important in itself because process intensification is meaningless if it is achieved at the expense of product recovery.
Combining multiple operations into a single step further compounds process intensification in a number of ways, beginning with reduction of the number of steps requiring development and validation. It also reduces the number of pieces of major equipment required for a process, which lowers capital costs, simplifies operator training and certification, and reduces the need to set up supply chains and maintain inventories of disposable media. It also reduces the number and/or volume of process buffers, tankage, and buffer storage space; it increases facility capacity by decreasing process time; and it reduces waste.
Not all purifications can be accomplished with one or even two chromatography steps, but the same principles do apply, and the opportunities for intensification increase. The tactic of dilute-to-direct-load on monoliths equally serves the need to reequilibrate buffers between purification steps, and it has been used effectively to do so in many influenza purification processes (4, 11–13). Whatever the number of process steps a purification process might include, pursuing a strategy of single-use compounds the overall degree of intensification.
References
1 Influenza (Seasonal). World Health Organization (WHO: Geneva, Switzerland, 2018); www.who.int/en/news-room/fact-sheets/detail/influenza-(seasonal).
2 Mischler R, Metcalf IC. Inflexal V a Trivalent Virosome Subunit Influenza Vaccine: Production. Vaccine 20(5s): B17–23; doi:10.1038/sj.gt.3302624.
3 Morenweiser R. Downstream Processing of Viral Vectors and Vaccines. Gene Ther. 12(1): S103–S110.
4 Fischer L, Wolff M, Reichl U. Purification of Cell Culture−Derived Influenza A Virus Via Continuous Anion Exchange Chromatography on Monoliths. Vaccine 36(22) 2018: 3153–3160; doi:10.1016/j.vaccine.2017.06.086.
5 Besnard L, et al. Clarification of Vaccines: An Overview of Filter Based Technology Trends and Best Practices. Biotechnol. Adv. 34(1) 2016: 1–13; doi:10.1016/j.biotechadv.2015.11.005.
6 Negrete A, Pai A, Shiloach J. Use of Hollow Fiber Tangential Flow Filtration for the Recovery and Concentration of HIV Virus-Like Particles Produced in Insect Cells. J. Virol. Met. 195, 2014: 240–246; doi:10.1016/j.jviromet.2013.10.017.
7 Gagnon P. The Emerging Generation of Chromatography Tools for Virus Purification. BioProcess Int. 6(6) 2008: S24–S30.
8 Kramberger P, Urbas L, Štrancar A. Downstream Processing and Chromatography Based Analytical Methods for Production of Vaccines, Gene Therapy Vectors, and Bacteriophages. Hum. Vaccin. Immunother. 11(4) 2015: 1010–1021; doi: 10.1080/21645515.2015.1009817.
9 Jindra C, et al. Attenuated Recombinant Influenza A Virus Expressing HPV16 E6 and E7 As a Novel Therapeutic Vaccine Approach. PLoS One 10(9s) 2015; doi:10.1371/journal.pone.0138722.
10 Oliver D. Virus Interactions with Membrane Filters. Biotechnol. Bioeng. 10(6) 1968: 877–889; https://doi.org/10.1002/bit.260100612.
11 Weigel T, et al. A Flow-Through Chromatography Process for Influenza A and B Virus Purification. J. Virol. Methods. 207 (2014): 45–53.
12 Transfiguracion J, et al. Particle Quantification of Influenza Viruses By High Performance Liquid Chromatography. Vaccine 33(1) 2015: 78–84; doi:10.1016/j.vaccine.2014.11.027.
13 Banjac M, et al. Purification of Vero Cell Derived Live Replication Deficient Influenza A and B Virus By Ion Exchange Monolith Chromatography. Vaccine 32(21) 2014: 2487–2492; doi:10.1016/j.vaccine.2014.02.086.
Mojca Tajnik Sbaizero is project manager at BIA Separations in Ajdovscina, Slovenia. Markus Wolschek is CSO, Manfred Reiter is CTO, and Thomas Muster is CEO of BlueSky Vaccines in Vienna, Austria. Corresponding author Pete Gagnon is CSO and Aleš Štrancar is CEO of BIA Separations in Ajdovscina, Slovenia, BIA Separations d.o.o., Mirce 21, 5270 Ajdovščina, Slovenia; Pete.Gagnon@biaseparations.com.