
An example of a combination product design: Novo Nordisk’s NordiPen growth-hormone injection device for children (WWW.NOVONORDISK.COM)
On 26 January 2015, CASSS hosted a program in its ongoing series of semiannual Chemistry, Manufacturing, and Controls (CMC) Strategy Forums at the Mayflower Hotel in Washington, DC. Since this series’s inception in 2002, each installment has focused on one of a wide array of topics spanning the fields of biopharmaceutical product development, manufacturing, analysis, quality, and regulation. For this forum, the program committee chose to devote a full program to a topic that was previously the focus of an afternoon session at their CMC Strategy Forum in July 2012: combination products (1).
In just the two-and-a-half years between the two programs, a great deal had transpired in the development and regulation of combination products. The US Food and Drug Administration (FDA) issued several guidance documents on topics such as pen and jet injectors and glass syringes (2, 3). Over this period, biomanufacturers gained more experience with design controls and implementing human factors guidances as well as added learnings from FDA interactions. Notably, the January 2015 forum took place after a significant milestone: Just 18 months earlier, the long-anticipated final rule in 21 CFR Part 4 Current Good Manufacturing Practice Requirements for Combination Products (4) had become effective. In a fitting bit of serendipity, the FDA released its draft guidance on the final rule just days before the forum convened in Washington (5).
| CMC Forum Series |
| The CMC Strategy Forum series provides a venue for biotechnology and biological product discussion. These meetings focus on relevant chemistry, manufacturing, and controls (CMC) issues throughout the lifecycle of such products and thereby foster collaborative technical and regulatory interaction. The Forum strives to share information with regulatory agencies to assist them in merging good scientific and regulatory practices. Outcomes of the Forum meetings are published in this peer-reviewed journal to help assure that biopharmaceutical products manufactured in a regulated environment will continue to be safe and efficacious. The CMC Strategy Forum is organized by CASSS, an International Separation Science Society (formerly the California Separation Science Society), and is supported by the US Food and Drug Administration (FDA). |
Overview
The purpose of this forum was to promote a better understanding of both regulatory and practical aspects of developing prefilled syringes, autoinjector pens, pumps, and other novel drug–device and biologic–device combinations to facilitate the transition from clinical development to licensure and beyond. (For the purposes of this paper, the term drug is intended to encompass both small-molecule pharmaceuticals and biological products.) Topics included design controls, design verification and validation, the role of human factors in combination product development strategy, regulatory expectations for combination product reviews and inspections, and best practices in early combination product development compared with that of postapproval and legacy products.
Early Development of Combination Products
Mark Lee of the FDA’s Center for Biologics Evaluation and Research (CBER) and John Towns of Eli Lilly chaired the morning session. Three presenters addressed different aspects of the early stages of combination product development, including novel drug–device combinations and human-factors studies. They highlighted the processes within and expectations of the FDA and other regulatory authorities, and they outlined industry experiences with combination-product development and work with regulators and contract manufacturers.
Lana Shiu of FDA’s Center for Devices and Radiological Health (CDRH) provided a regulator’s perspective on combination products. As a senior medical advisor in CDRH’s Office of Device Evaluation (ODE) and drawing from her experience as both an engineer and a physician, Shiu illustrated the regulatory mechanisms, pitfalls, and best practices that sponsors should consider when developing and submitting a combination product for review.
The Office of Combination Products (OCP) determines which FDA center should be assigned the lead for a combination product based on each product’s primary mode of action (PMOA). Products whose PMOA is achieved by a device constituent with a drug serving a secondary action (e.g., a drug-eluting stent) are regulated by CDRH under device provisions. But for most biopharmaceutical products comprising a novel biologic agent filled into a device (e.g., syringes or autoinjectors), one of the FDA’s two drug review centers — CBER or the Center for Drug Evaluation Research (CDER) — typically will be assigned as the lead center because the drug itself is responsible for the product’s PMOA.
When CDER or CBER serves as the lead center for review of a combination product, CDRH/ODE reviews all engineering aspects of the device in collaboration with the lead center review team. In particular, the General Hospital Devices Branch within ODE’s Division of Anesthesiology, General Hospital, Respiratory, Infection Control, and Dental Devices (DAGRID) fields the vast majority of CDER and CBER review requests for combination products. Such products include prefilled syringes, autoinjectors, jet injectors, wearable pumps, and implanted ports.
Shiu provided some historical context for CDRH’s increased oversight of prefilled syringe combination products in recent years. Throughout 2010–2013, mechanical incompatibilities were reported when some needleless prefilled glass syringes were used with intravenous (IV) lines containing needleless connectors in critical-care settings. Those incompatibilities caused syringe breakage and an inability to administer time-critical medications. Slight differences in nozzle dimensions and manufacturing tolerances allowed by ISO standards were not critical when syringes were used with a needle for manual injection, but those issues became highly critical for needleless IV connectors for IV push medications. Those incidents led to a greater appreciation for the need to review and regulate prefilled syringes as combination products as well as the issuance of a specific draft guidance (3).
Shiu summarized by stating that a prefilled syringe needs to be designed and developed in a manner suitable for its intended use just like any other combination product. Mere adherence to standards or use of previously approved components is no substitute for demonstrating functionality of a combination product and compatibility of a drug. End-user human factors also must be considered. Shiu advised that companies wishing to fill a drug into a previously cleared device under 510(k) submission should be mindful that drug-specific data often are needed to demonstrate stability and compatibility of a final combination product. Such analyses include extractables and leachables studies and drug-adsorption–delivered-dose studies.
Shiu touched on ODE’s experiences in reviewing human factors for combination products and stressed the importance of considering the intended-user population of a final product. If an established platform device is to be used with another drug for a different environment of use or end-user population (e.g., switching from healthcare professionals to patient self dosing or applying an available in-house autoinjector design to a different disease), new human-factors studies usually are needed. Those studies can be waived specifically by FDA based on detailed prior discussion. Sponsors also need to consider other medications that patients are likely to be taking and ensure that their combination-product designs minimize risk of confusion and medication errors.
Kristi Kistner (Amgen) presented her company’s experiences in early collaboration with FDA. Engaging the FDA while a combination-product program is still in its early clinical stages gives companies crucial input around which to build their development plans. It provides the agency with a line of sight into innovation and allows it an opportunity to identify critical issues and convene reviewers with the appropriate expertise.
That kind of early outreach is crucial to overcoming inherent limitations of combination product review processes. Although drugs contained in prefilled syringes, autoinjectors, and pumps are assigned to CDER or CBER as the lead center based on a product’s PMOAs, it is CDRH that houses the FDA’s subject matter experts in device engineering, design, and validation. Often, review of the device constituent at CDRH might not be initiated in the early stages of development. Thus, if a company does not make a proactive effort to engage CDRH experts during phase 1, sponsors run the risk of encountering device-related issues in later stages of development, resulting in potential misalignment and costly course corrections. Kistner noted that even under the best circumstances, challenges can arise during review because of differences between review divisions of CDER and CBER regarding requirements or expectations for drug-specific data at the intersection of the drug and device components of a combination product.
Kistner explained that intrinsic differences between drug and device development time lines are further reasons for engaging early with regulators. For the drug constituent of a combination product, regulatory CMC guidance provided by the FDA at an end of phase 2 (EOP2) meeting often is actionable in time for phase 3 trials, which may be initiated six to nine months postmeeting. For the device constituent, the path from critical design inputs to a phase 3–ready device has a much longer time horizon: from one to four years. Thus, waiting until an EOP2 meeting to solicit FDA feedback about a device design will come too late to incorporate any learnings into a commercial-equivalent device and still keep phase 3 clinical studies on schedule.
Companies therefore should make the most of opportunities to touch base with FDA centers and the Office of Combination Products (OCP) to ensure that device development keeps pace with drug development. Indirect interactions with agency representatives through industry consortia (e.g., Combination Products Coalition) and standards organizations (e.g., Association for the Advancement of Medical Instrumentation, International Organization for Standardization, and the International Electrotechnical Commission) can be just as valuable as formal product-specific meetings.
Sujit Basu (Shire) presented on the unique challenges of developing combination products for rare diseases. Development programs with potential to address significant unmet medical needs for serious diseases often are candidates for accelerated development. The decision to expedite development is driven by clinical outcomes and clinical strategy. And the inevitable compression of CMC timelines means that careful thought must go into combination product plans from the very beginning. Basu emphasized that with less time during and between clinical phases to develop and optimize a product, project teams must drive continuously toward understanding the relationship among drug structure, drug function, formulation, manufacturing process, device-design requirements, human factors engineering, and drug–device compatibility. Development team members need to share a common awareness that a change in one of those aspects of a combination product can have ramifications for others, and thus cross-functional alignment is especially critical to a product’s success.
Basu presented examples of development programs for lysosomal storage diseases that necessitated the use of an implantable device for intrathecal drug delivery. The final combination product presentation and its method of use must be considered in the design of a drug product constituent, and vice versa.
A drug’s stability requirements and dosage form drive formulation decisions, which can influence its critical quality attributes (CQAs). But changing a drug’s route of administration from intravenous to intrathecal, for example, can alter its risk profile and thus the degree of criticality of CQAs themselves. In-use studies also are crucial for demonstrating that drug-product quality is maintained during the administration process and that device components support drug stability.
Basu shared excerpts from a risk assessment to identify potential failure modes for a device. He emphasized the importance of viewing risk within the context of a disease state and intended patient population. For example, implantable intrathecal ports had previously been used to deliver pain medications in advanced-stage cancer patients. The patient population presents a very different device stress and physical activity profile than that of pediatric patients with lysosomal storage diseases. Once physical forces and failure modes were identified, mechanical test methods were developed to test for device breakage under clinically relevant conditions of children’s body movement and external trauma. That example of a novel, implantable combination product was intended to highlight the importance of using an integrated quality approach that incorporates clinical considerations for the intended drug–device combination from an early stage of development.
The morning’s presentations were followed by a panel question-and-answer session. The speakers were joined by three additional panelists: Martin Nemec of Health Canada’s Centre for Evaluation of Radiopharmaceuticals and Biotherapeutics (CERB), Irene Chan of FDA/CDER’s Division of Medication Error Prevention and Analysis (DMEPA), and Molly Story of Sanofi.
Managing Device Design Changes During Development: One important element of early phase device development is the need to manage changes carefully during clinical development. Panelists were asked to reflect on what types of technical changes to a device might affect interpretation of clinical data, how to mitigate those changes, and lessons learned from combination products already on the market.
Both FDA and industry panelists noted that the degree of supporting data should be commensurate with change made to a device. Some design changes may warrant additional human-factors studies, pharmacokinetics data, or (for certain changes to autoinjector pens) studies to demonstrate that a postchange design can deliver drug to the same tissue plane as a prechange device. Studies to evaluate potential changes in pain upon injection should be considered when a drug product is migrated from one device to another (e.g., from prefilled syringe to autoinjector). However, design changes that can be considered minor or those that are limited solely to one aspect of a combination-product use experience (e.g., alterations to an autoinjector firing mechanism without any other changes) might require only those studies that are relevant to a given change, such as human factors. When proposing a device design change to the FDA, companies should present a risk assessment and the rationale for why certain supporting studies are necessary or unnecessary. FDA reviewers are willing to consider such proposals.
Panelists suggested that companies should think about the inherent limitations of human-factors studies as they are currently conducted. Sponsors ought to consider the extent to which a person in an office injecting a placebo-filled device into a pad truly represents the experience of a patient using that same device to inject him- or herself with drug at home. Several types of human-factors studies can be valuable in validating particular device design changes. Biomanufacturers have an opportunity to conduct the right studies to learn the right information to support a given change.
An increasing number of biomanufacturers have been interested in understanding “actual use” and “real-life patient handling” of self-administered combination products. Companies that wish to probe such questions find that they may receive varied feedback from different review divisions at the FDA. But often that is due to disease-specific considerations. Critical factors in combination-product use can be quite different between patients with rheumatoid arthritis and those with diabetes, for example.
Prefilled Syringes As Combination Products: Health Canada’s approach to classification and review of combination products is analogous to that of the US FDA: Most prefilled syringe products are reviewed by the drug directorates on the basis of their PMOAs. For devices previously established in Canada and relatively straightforward in design, consultation with medical device reviewers can be minimal. That is because drug reviewers are fully capable of considering questions of extractables and leachables, lubricant use and application, syringe manufacturing and components, and so forth. The FDA panelists pointed out that sponsors that manufacture drugs in prefilled syringes are considered to be combination-product manufacturers — even if not assembling them into more complex delivery devices. Such companies are expected to adhere to combination-product current good manufacturing practices (CGMPs) according to 21 CFR Part 4 (a topic discussed in greater detail during the afternoon session).
Biomanufacturers with core expertise and systems oriented toward drug CGMPs may partner with other companies for prefilled syringe design and manufacturing needs. But as the holder of a biologics license application (BLA) or new drug application (NDA), a sponsor is ultimately responsible for ensuring good product quality and compliance of a combination product as a whole, not just for the drug constituent. Drug companies should leverage the knowledge of their device manufacturing partners to ensure good product quality. The FDA panelists added that sponsors should consider bringing their device partners to the table when discussing specific review matters, especially if answers to reviewers’ questions reside in a drug master file (DMF) containing confidential device-related information that sponsors themselves are not authorized to see.
Global Development of Combination Products: Panelists discussed considerations of global development for combination products and explored the differences in submission and review between the United States and European Union. Currently, the European Medicines Agency (EMA) submissions of device CMC information for combination products are considerably shorter than those typically provided to the FDA. That can lead to unintended divergence in how risks are managed between the regions.
In one example, the FDA deemed user-education materials to be an essential part of risk mitigation. So those materials were filed and reviewed with the combination-product submission. But when the same product was submitted to the EMA for review, those materials were considered to be optional and did not need to be filed. In another example, the EMA provided feedback that certain human-factors information provided in a submission was insufficient. The sponsor actually had the necessary information captured in a report. But because of differences in CMC content of combination product submissions, those data were provided upfront to the FDA but not to the EMA. The prevailing sense among the panelists was that the overall data package ultimately given to the FDA and EMA ends up being similar. But significant regional differences remain in how such information is presented and when during the review cycle it is provided.
Developing Platform Devices: The development of platform autoinjectors for multiple products in a company’s portfolio can be an efficient means to fulfill patient needs for convenience. But panelists discussed the caveats that biopharmaceutical companies must bear in mind in such cases. Physical properties of different drug solutions and their influence on device functionality can be critical. Platform devices need to be designed and demonstrated to accommodate the full intended range of viscosities, forces, volumes, delivery speeds, and needle lengths that will be used. Echoing a point from an earlier presentation, just because a prefilled syringe meets certain standards does not necessarily mean that it is suitable for use with a given autoinjector, nor is it automatically suitable for use across different drugs from the same sponsor.
Biomanufacturers that are developing a platform autoinjector or pump should consider the ramifications of which US regulatory pathway they choose to follow. If a device is filed as a combination product with a drug under an NDA or BLA, the device is afforded seven years of exclusivity because of its linkage to the drug. By contrast, filing under Section 510(k) designates the device for general use, and other companies may develop similar devices that nominally achieve the same function. Regardless of submission pathway or lead center, both the drug and device constituents will be reviewed accordingly by CDER/CBER and CDRH. Sponsors are strongly discouraged from submitting two separate applications (as some have done in hopes of expediting drug and device review). That serves to create a “shared nightmare” between centers and will delay approval.
Regulatory Submissions and Correspondence: Both industry and agency representatives shared their best practices for regulatory correspondence and creating quality submissions. The FDA advised companies to be clear and succinct in writing the device sections of a submission. For complex combination products, photos and videos can be very helpful for reviewers to gain a better understanding of a device’s operation and function. In early interactions with health authorities during development, sponsors should provide background and context for the questions they wish to ask of regulators, followed by explicit proposed solutions that will prompt concrete feedback from device reviewers.
Premarket, Postapproval, and Legacy-Product CGMP Considerations
Jee Chung (FDA’s CDER) and Gary Hartman (Amgen) chaired the afternoon session. Presenters focused on later stages of a combination product’s lifecycle and highlighted additional areas of focus of FDA reviewers and investigators. They also provided thoughts regarding industry best practices.
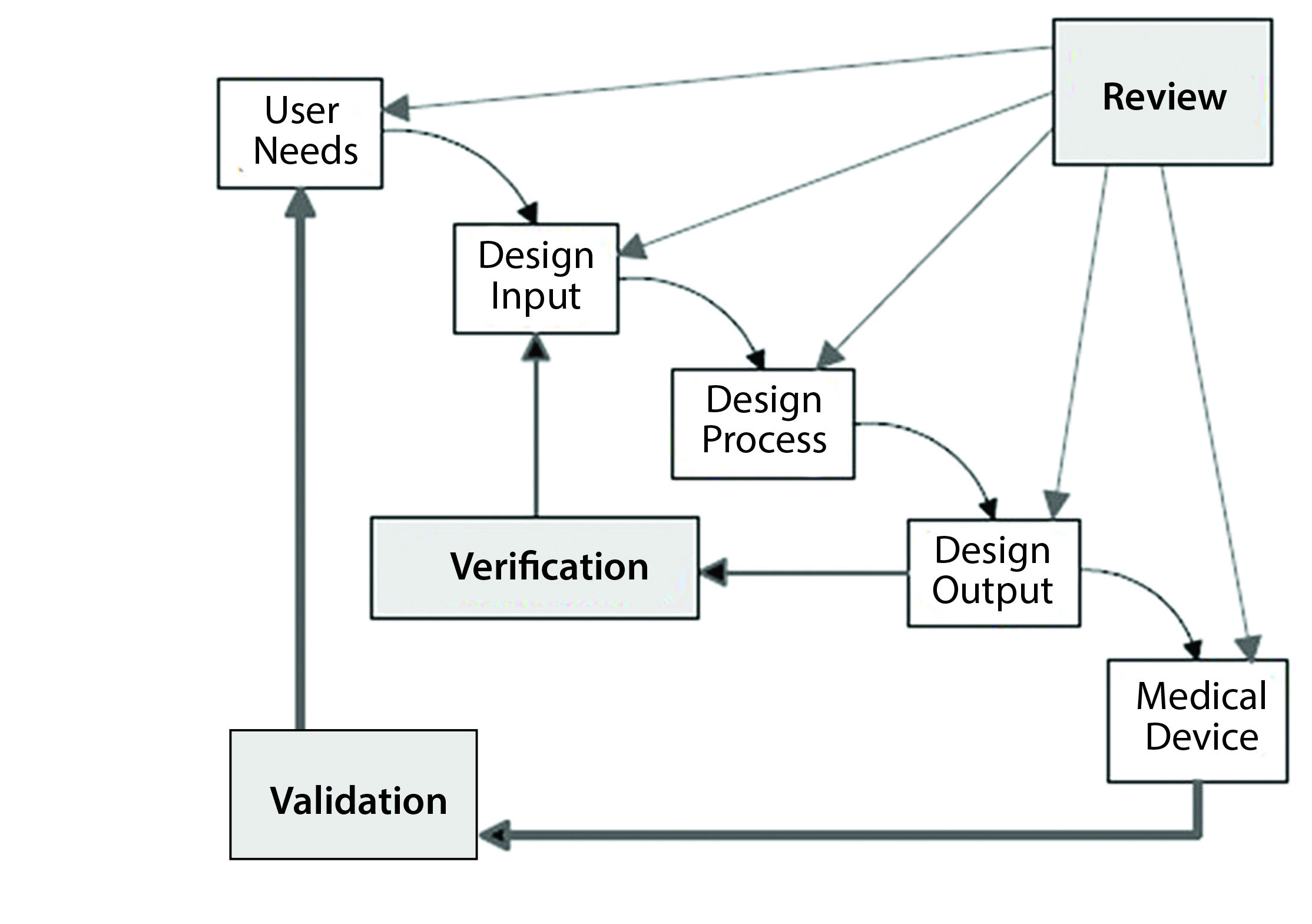
Figure 1: The design control process for drug–device combination products (Reprinted with permission of Health Canada)
Steven Badelt (Suttons Creek) spoke about best practices in design verification and validation from a systems engineering perspective. By definition, a combination product is a system. Systems engineering encompasses numerous activities that are part of the regulations for combination product development (Figure 1), including identification of customer needs and product functionality, documentation, design inputs and outputs, and validation (6).
Systems engineering can integrate the disciplines that will work on the entire system throughout development. In this paradigm, combination products can be broken down into subsystems in which the drug is not “king.” Instead, the drug is treated as one component of an entire system that is required to achieve user needs, and the entire system must be fully designed and evaluated.
One key aspect noted is the need for frequent communication among engineering and regulatory affairs and all other groups working on a project.
Another best practice is setting up document trees at the planning stage to ensure that product documentation includes the entire story of planning, design, implementation, verification, and validation. A design should start with clear and specific requirements for a given system. Those are important for postapproval changes. Although including requirements that are “to be determined” are acceptable, the processes by which those requirements will be defined must be clearly documented.
Many verification and validation complications start with poor requirement identification at early stages or an incomplete understanding of differences between verification (confirmation that specified requirements have been fulfilled) and validation (confirmation that the particular requirements for a specific intended use can be specifically fulfilled) (6). Vendor specifications should not be used as system requirements. Requirements for a system should be determined, and then an appropriate vendor or component should be selected based on those requirements. Performing system modeling can help biomanufacturers identify holes in system requirements. Specifying system verification and validation plans and protocols when documenting system requirements can provide a better understanding of an entire program.
Verification does not need to be overly cumbersome. For example, certain design aspects can be verified, through inspection rather than with specific testing. In addition to predetermined acceptance criteria and sample sizes, protocols should take into account materials traceability, requirement tags, and standards references. Although standards-driven verification is suggested, it is important to not follow standards blindly but to consider risk profiles of combination products when generating verification protocols. Reports should be comprehensive and include deviations and discrepancies. Useful information and tools for further consideration can be found in the International Council on Systems Engineering (INCOSE) systems engineering handbook (7).
The FDA CDRH Office of Compliance (OC) evaluates compliance with applicable regulations specified in 21 CFR 820. In her presentation, Isabel Tejero (FDA/CDRH/OC) described current expectations. She explained that 21 CFR Part 4 did not introduce new requirements, but it provides clarification of ways to meet required GMP obligations using a more streamlined approach (4). In addition to drug CGMP requirements outlined in 21 CFR 210 and 211, drug manufacturers must comply with device CGMP obligations listed (see box “21 CFR 4.4(b)(1)”). Some of those requirements already are familiar to drug manufacturers: e.g., management responsibility, purchasing controls, and corrective and preventive actions (CAPA). And some requirements are applicable only to specific devices (e.g., installation and servicing).
| 21 CFR 4.4(b)(1): A Streamlined Approach |
To ensure quality of drug–device combination products, biomanufacturers must meet drug CGMPs (21 CFR 210 and 211) and selected provisions of device quality system regulations, including
|
Tejero pointed out that design controls requirements under 21 CFR 820.30 should warrant the attention of a drug manufacturer’s development and quality organizations. Such requirements are not retroactive for products marketed before 1 June 1997, but they are applicable to products changed after that date. The stage at which design controls are required to begin is not explicitly defined by regulation. The timepoint at which a manufacturer commits to develop a product (when a product moves from research only to development) is considered the general point at which design controls become applicable.
Tejero also highlighted the importance of considering whole products, not only constituent parts, when addressing design requirements and during documentation and communication. A design history file (DHF) should include a comprehensive list or spreadsheet that references all documentation that supports the design, manufacturing, and evaluation of a combination product, including both device and drug constituents. Responsibilities of different groups and the mechanisms through which they interface should be clear. That allows manufacturers to determine the degree to which a design process is integrated.
Design inputs should include an organized mechanism for addressing incomplete, ambiguous, or conflicting requirements. While design outputs are being established, testing and manufacturing should be considered. Manufacturers should include independent observers to participate in design reviews. They often can identify potential issues that are overlooked by staff who have been directly involved in projects. Although regulations specify only risk analysis as part of validation, both it and risk management should be applied throughout a combination product’s lifecycle. All changes that are made after design review and approval of initial design inputs must be documented. Manufacturers must ensure that design requirements are still met and that safety and effectiveness of their products are not negatively affected.
Products should be brought into compliance with 21 CFR 820 using the CAPA system. Manufacturers should perform a systematic evaluation of all combination products, and retrospective DHFs should be generated. Those DHFs should be as complete as possible; however, the FDA has recognized that some elements cannot be recreated (e.g., meetings that occurred in the past). Risk assessments should be used to evaluate items that cannot be recreated, and control activities should be used to fulfill requirements that cannot be addressed and to mitigate identified risks. All activities performed to meet the current requirements should be carefully documented. Additional guidance can be found in the draft FDA guidance on CGMP for combination products (5).
Sean Creighton (FDA’s Office of Regulatory Affairs, ORA) provided his insights into the mind of an FDA medical device national expert investigator. Based on his participation in numerous inspections, Creighton shared advice for preventing medical device observations. Manufacturers should oversee processes surrounding combination products to identify and address problems before they are identified by FDA investigators. Manufacturers should demonstrate control of their quality systems.
To help ensure compliance with regulations, a system should be user friendly. In addition, employee education should convey a position of ownership of regulations related to their responsibilities. The preambles to regulations are useful educational tools because they explain why regulations were implemented. Manufacturers may find it useful to commingle global regulations to provide employees with a complete picture of regulations and resources available. Work instructions, protocols, and other systems used by staff should be sufficiently complete and clear to help them meet applicable regulations. Employees need to understand what a design change is so that changes are appropriately evaluated, documented, and reported to regulators.
With respect to design controls, Creighton suggested that it can be useful to develop a matrix or spreadsheet with links between design inputs and outputs and among verification, validation, and risk assessment activities used to evaluate products. That creates a roadmap for product development. One of the most common observations identified is a lack of predetermined acceptance criteria for design verification as well as a lack of adequate sample sizes. Typically, test methods need to be created for verification testing. Such methods must be validated and often transferred to manufacturing organizations.
Creighton noted that manufacturers’ CAPA systems frequently have been the subject of FDA observations upon inspection. The causes of all nonconformances should be investigated and addressed in a timely manner. Information and data should be gathered and tracked until an investigation is complete. Although the “big picture” should be evaluated, specific problems also need to be identified and resolved. Effectiveness checks for CAPAs should include quantifiable metrics and ensure that a correction does not initiate a different problem.
For the afternoon panel discussion session, Badelt and Tejero were joined by Donna French of Genentech (a member of the Roche group), Irene Chan of FDA’s CDER, Cathy Parker of Health Canada, and Anthony Watson of Biogen.
Legacy Products and Leveraging Cross-Product Data: Risk assessments should be used to evaluate deficiencies in design controls of legacy products and be product specific. It might be possible to use reviews of complaint data and other market data to fulfill or partly fulfill some design control requirements. But additional studies also might be necessary.
Manufacturers can leverage data from other products, but the appropriateness of doing so must be carefully considered. It is important to understand your product in the context of its situation of use and the risk associated with its different aspects (e.g., missed or partial doses). Interactions between a device and different drugs and potential differences in user populations can preclude leveraging cross-product data for some combination-product characteristics. However with such caveats in mind, molecule-independent aspects can be leveraged as part of design verification, and manufacturers might be able to use a single assessment to evaluate use across a broad range of users. Then they can apply those data to many uses of a device or use data derived from a population with similar critical characteristics. Manufacturers should provide regulators with their rationale for conducting additional studies to fulfill design requirements. Additional collaboration among industry can be a potential mechanism for supporting leveraging of cross-cutting data.
Device Inspection: The FDA considers the comprehensive inspection documentation when determining inspection outcomes. The agency evaluates inspection observations (reported on FDA Form 483), inspection reports, and responses to the 483 observations submitted by manufacturers. If design-control gaps are closed before inspection, a manufacturer would be in compliance at the time of inspection, and such gaps should not be observations. Evaluation of inspections is harmonized at the FDA Center, and final assessment depends on a remediation plan. The agency does not expect everything to be fixed in one day, but it is important to begin risk assessments and remediation of gaps (e.g., in DHFs) early. For all combination products, it is important to remember that a risk assessment is a living document. There must be mechanisms for adding newly identified risks to the assessment.
Regulatory Submissions for the United States and Canada: Requirements for combination products regulated by Health Canada do include some differences with respect to US requirements predominantly discussed during this forum. The reporting categories for changes to such products are classified based on significance. Specific guidance includes a flow chart for design changes and the types of submissions to file in Canada (8). For combination products submitted to the FDA as BLAs, documentation that addresses the 4.4(b)(1) requirements should be included in the common technical document (CTD) section 3.2.P.7 (container–closure). The PMA submission guidance can be referenced for guidance regarding information to include (9).
Although most historical experience for biopharmaceutical combination products (and the focus of this forum) largely focused on prefilled syringes and autoinjectors, novel technologies are increasingly being regulated as combination products. Regardless of the device technology being used, a number of common themes emerged from the day’s presentations and discussions. Such topics included the importance of communication, documentation, considerations for a combination product as a whole (rather than only individual components), verification and validation plans at the time requirements are identified, careful evaluation of proposed human-factors studies, and use of strong risk analyses.
Effective and early communication among manufacturers, regulators, engineering/product development groups, regulatory affairs, and other groups interfacing on a project is essential for efficient product development. All aspects of product development should be thoroughly documented, and the DHF should be comprehensive. Identification of system requirements is a critical aspect of product design. Such requirements should be unambiguous and specific to a product, patient population, and indication. Human-factors study development should include consideration of the characteristics of the specific intended patient population and the specific attributes of the drug component.
Manufacturers should perform consistent, product-specific risk analyses no matter how simple a product may appear. Risk analysis and risk management should be applied throughout the lifecyle of a combination product, and risk assessments and DHF should be updated as appropriate. Comprehensive design controls and documentation will support straightforward combination product review and related inspections.
| Disclaimer |
| The content of this manuscript reflects discussions that occurred during the CMC Strategy Forum. This document does not represent officially sanctioned FDA policy or opinions and should not be used in lieu of published FDA guidance documents, points-to-consider documents, or direct discussions with the agency. |
References
1 Mire-Sluis A, et al. Drug Products for Biological Medicines: Novel Delivery Devices, Challenging Formulations, and Combination Products, Part 2. BioProcess Int. 11(6) 2013: I11
2 Guidance for Industry and FDA Staff: Technical Considerations for Pen, Jet, and Related Injectors Intended for Use With Drugs and Biological Products. US Food and Drug Administration: Silver Spring, MD, June 2013; www.fda.gov/downloads/regulatoryinformation/guidances/ucm147095.pdf.
3 Guidance for Industry and FDA Staff: Glass Syringes for Delivering Drug and Biological Products: Technical Information to Supplement International Organization for Standardization (ISO) Standard 11040-4. US Food and Drug Administration: Silver Spring, MD, April 2013; www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM346181.pdf.
4 21 CFR Part 4: Current Good Manufacturing Practice Requirements for Combination Products. Revised 1 April 2016. US Food and Drug Administration: Silver Spring, MD; www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=4&showFR=1.
5 Guidance for Industry and FDA Staff: Current Good Manufacturing Practice Requirements for Combination Products. US Food and Drug Administration, Silver Spring, MD, January 2015; www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM429304.pdf.
6 Design Control Guidance for Medical Device Manufacturers. US Food and Drug Administration: Silver Spring, MD, March 1997; www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm070642.pdf.
7 Walden DD, et al., Eds. INCOSE Systems Engineering Handbook: A Guide for System Life Cycle Processes and Activities, 4th Edition. John Wiley & Sons: Hoboken, NJ, 2015.
8 Guidance on the Risk-Based Classification System for Non-In Vitro Diagnostic Devices (non-IVDDs). Health Products and Food Branch, Health Canada, June 2015; www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/md-im/applic-demande/guide-ld/gd_rbc_non_ivdd_lg_scr_autres_idiv-eng.pdf.
9 Guidance for Industry and FDA Staff: Quality System Information for Certain Premarket Application Reviews. US Food and Drug Administration: Silver Spring, MD, February 2003; www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm070899.pdf.
| Global Steering Committee for These Forums |
| Siddharth J. Advant (Celgene Corporation, USA), Daniela Cerqueria (ANVISA, Brasil), Yasuhiro Kishioka (PMDA, Japan), Junichi Koga (Daiichi Sankyo Co., Ltd., Japan), Steven Kozlowski (OBP, CDER, FDA, USA), Ingrid Markovic (CBER, FDA, USA), Rohin Mhatre (Biogen, USA), Anthony Mire-Sluis (AstraZeneca, USA), Wassim Nashabeh (F. Hoffmann-La Roche Ltd., Switzerland), Ilona Reischl (AGES/BASG, Austria), Anthony Ridgway (Health Canada, Canada), Nadine Ritter (Global Biotech Experts, LLC, USA); Mark Schenerman (MedImmune, USA), Thomas Schreitmueller (F. HoffmannLa Roche Ltd., Switzerland), Karin Sewerin (BioTech Development AB, Sweden) |
Siddharth Advant is currently at Celgene; at time of this forum, he was president of biologics at Kemwell Biopharma. Jee Chung is a biologist at CDER, FDA. Gary Hartman is director of quality assurance for combination products at Amgen. Mark H. Lee is currently associate group director at Genentech and was a biologist at FDA’s CBER at the time of the forum. Lana Shiu is director of regulatory at FDA’s CDRH. John Towns is senior research fellow, global regulatory affairs at Eli Lilly and Company. Sarah Kennett is review chief at OBP, OPQ, CDER, FDA. Andrew Weiskopf is director of regulatory affairs–CMC at Biogen.