
WWW.PHOTOS.COM
Precious patient samples, contamination concerns, and limited product purification options have compelled manufacturers of cellular immunotherapies (iTx) such as chimeric antigen receptor T cells (CAR-T) and T-cell receptor (TCR) technologies toward the disposables industry. Such companies are implementing single-use technologies (SUTs) almost exclusively (1). But despite the dominance of disposable bioprocess platforms and their extraordinary growth in the iTx marketplace, researchers have made limited efforts to understand the perennial and critical bioprocessing risks of leachables and extractables.
Here we outline the potential impact on iTx of leachables and extractables and discuss relevant regulations, guidelines, and risk-assessment approaches. Fortunately, biopharmaceutical producers of monoclonal antibodies (MAbs) and recombinant proteins have been investigating the consequences of leachables and extractables on manufacturing processes (1–3) for years. iTx companies (and the regenerative medicine market in general) can benefit from lessons learned over the past decade of commercial-scale biologics manufacturing in single-use platforms.
Unlike robust commercial cell lines used to produce biologics, current autologous iTx have relatively small volume requirements that contribute to increased sensitivity to culture conditions (4). Biomanufacturing process-fluid volumes also are affected by batch and lot sizes of allogeneic (universal donor, scale-up) and autologous (patient specific, scale-out) iTx. For example, a scale-out manufacturing process for an autologous therapy can increase the overall surface area available to affect leachables and extractables per volume of a cell culture because current standard rocking-motion bags used for expansion steps have relatively low-volume batch sizes. A high ratio of surface area to volume further strengthens the argument for in-depth analysis of the effects from leachables and extractables.
Regulatory Guidelines for Disposable Systems
Elastomeric and polymeric materials present the main source of leachables and extractables in SUTs. The presence of these additives provides product benefits such as increased stability, robustness, and antislip properties (3). Existing US Food and Drug Administration (FDA) and European Medicines Agency (EMA) regulations stipulate that production equipment should not “present any hazards to the product” and should not be “reactive, additive, or absorptive” (3–6).
The development, validation, and testing of disposables must be carried out under guidelines from the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) and the United States Pharmacopeia (USP) in a laboratory that complies with current good manufacturing practices (CGMP) (7). Industry experts recommend a thorough yet realistic approach to assess potential extractables. The strategy should test within a range of parameters such as organic and water-based solvents, reasonable pH extremes, and reasonable temperatures (2). Note that currently there are no published allowable limits or regulations available to establish a target for biomanufacturing processes (7). The amount of allowable leachables and extractables (as it concerns regulatory affairs) is determined only when it is shown to influence product quality, production consistency, product stability, and/or patient safety.
A number of industry organizations, nongovernmental agencies, and consortiums have worked together to provide industry guidance and best practices. In the United States and other English-speaking countries, the Bio-Process Systems Alliance (BPSA) works to encourage the adoption of single-use manufacturing technologies. Its members share best practices on topics such as extractables and leachables testing (8, 9). In spring 2010, a temporary working group of German-speaking countries took the name of “Single Use Technology in Biopharmaceutical Production” formed from the Bioprocess Technology working committee of DECHEMA (10). Both BPSA and DECHEMA highly recommend that extractables and leachables testing programs be implemented as early in a development process as possible to reduce burdensome late-stage process changes. An effective program is based on a thorough understanding of an entire process, including “R&D studies, process descriptions, batch records, standard operating procedures (SOPs), technical reports, batch testing, data trending, and operation parameters” (3).
The list below outlines common user-defined problems with the introduction of SUTs into a production process. Many of these issues also complicate the ability of manufacturing groups to assess appropriate leachable and extractable guidelines for their processes (10):
- The industry lacks defined, pharmaceutical-grade polymers.
- The qualification and validation data of all producers are variously compiled and informative, but the data often do not list all additives.
- The industry has no regulatory requirements for SUTs. Some specific single-use parts are listed in the requirements, but such requirements are not defined for all production steps.
- In general, few studies have been conducted on risk analyses of SUTs.
- Requirements for SUTs can vary widely based on a number of different factors, including duration of product contact with SUTs and the importance of specific extractants.
- The industry has no evaluated analytical methods with acceptance criteria for leachables and extractables.
Potential Negative Effects of Leachables and Extractables
To establish a baseline for guidance about cellular immunotherapy manufacturing concerns that pertain to leachables and extractables, it is helpful to evaluate a typical biomanufacturing process for MAbs or recombinant proteins (3) (Figure 1).
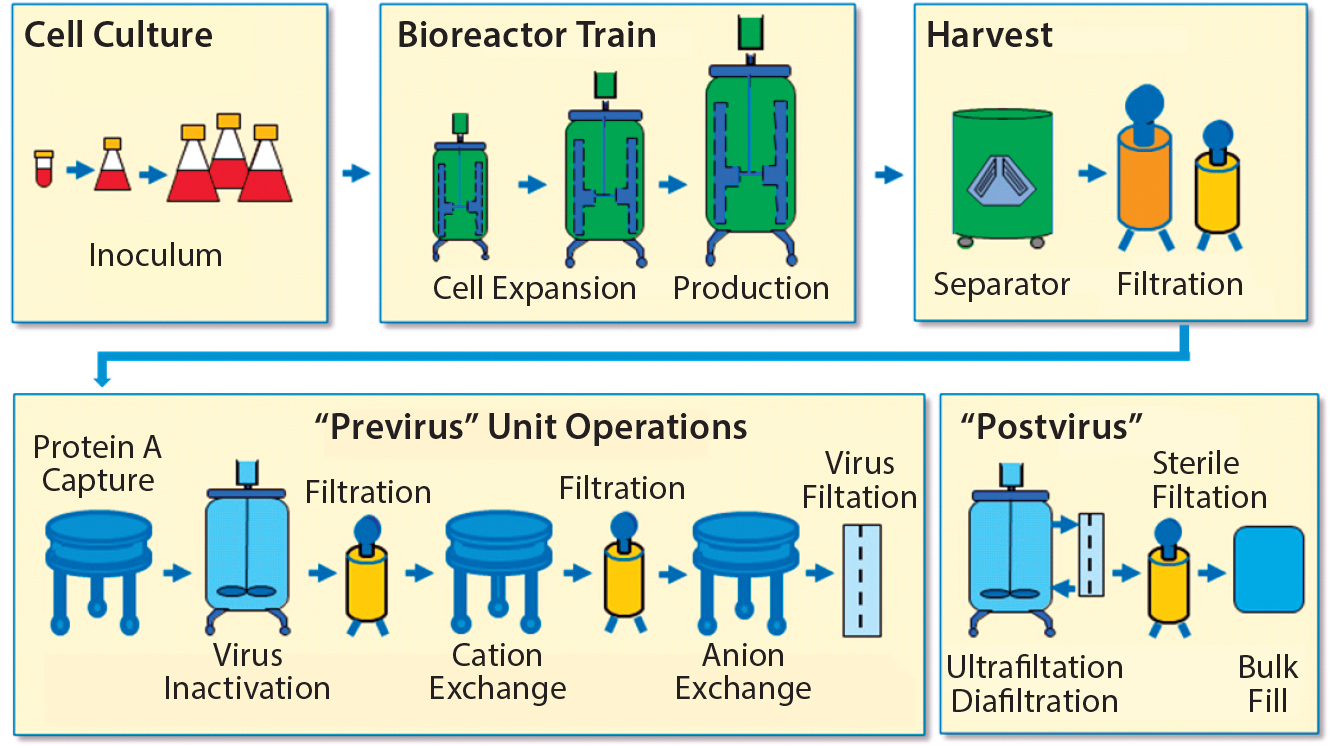
Figure 1: Monoclonal antibody biomanufacturing process (15)
Leachables and extractables can be present in almost every unit operation of a biomanufacturing process that uses SUTs. Leachables can be introduced through interactions and contact time with different materials and additives used to construct films, bags, tubing, filters, and other process materials. Also of concern are the leachables and extractables present in holding tanks for buffers, media, water for injection, and other liquids required for manufacturing processes. Unwanted contaminants can accumulate from each step and must be evaluated.
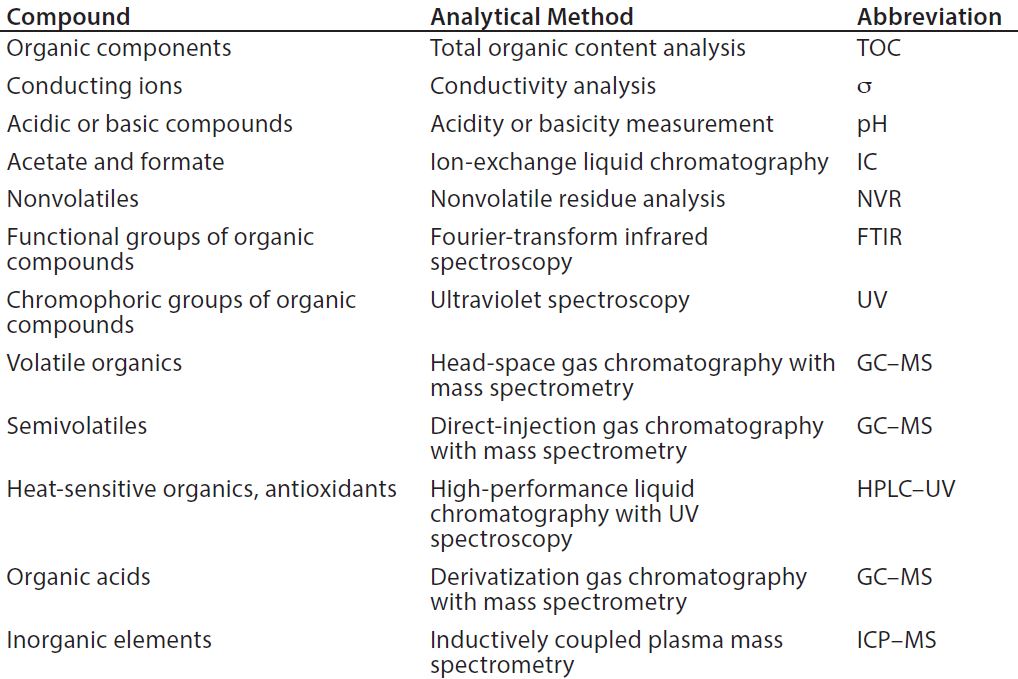
Table 1: Common leachable and extractables compounds (13)
To determine the appropriate levels of leachables and extractables allowed to be present in a final drug product, manufacturers must carry out a thorough risk-based assessment (3). A comprehensive risk assessment will rely on detailed process knowledge, analytical expertise, and available equipment, to analyze for common leachables and extractables compounds (11) (Table 1).
Multiple factors can affect the overall risk of biopharmaceutical processes associated with the effects of leachables and extractables. These are generally divided into two groups: process-related factors and dose-related factors.
| Extractables and Leachables |
| Extractables: Chemical compounds that migrate from any product-contact material when exposed to an appropriate solvent under exaggerated conditions of time and temperature (5). |
| Leachables: Chemical compounds (typically a subset of extractables) that migrate into a drug formulation from a product-contact material (e.g., elastomeric, plastic, glass, stainless steel, and coating components) as a result of direct contact with a drug formulation under normal process conditions or accelerated storage conditions and are found in a final drug product (5). |
| Regulations: 21 CFR 211.65 (a), 21 CFR 600.11 (b), ICH Q7 (1–3), 21 CRF 211.94 (a), 21 CFR 600.11 (h), 21 CFR 600.3 (b) (8), reference 2 (21 CFR volume 7), and reference 6. The US Federal Food, Drug, and Cosmetic Act requires additional control of leachables and extractables, as referred to under “adulterated products” (2). |
Process-related factors include contact area per volume of drug product and length of contact time a drug may have with various materials. Some cytotoxic compounds such as bis(2,4-di-tert-butylphenyl) phosphate (bDtBPP) (12) have been found to leach from single-use films used in culture and media bags. That specific compound was responsible for reduced cell growth in a Chinese hamster ovary (CHO) process and inconsistent cell viability due to varying leachable amounts per bag per lot, even from the same manufacturer. bDtBPP is formed from the degradation of a common antioxidant stabilizer, which is added to many flexible polymers such as those used in biomanufacturing (12).
Dose-related concerns take into account factors such as dosage volume and dosage frequency. For instance, a specific allowable leachable concentration for a given compound may be relatively high for a one-time, low-volume dose (e.g., a 2-mL injection) compared with a high-volume dose (e.g., 2,000-mL transfusion). Therefore, the overall accumulation of that leachable in a patient must be assessed by taking into account total dosage amount (3). In the above example, the allowable leachable concentration for the 2-mL injectable could be 1,000 times higher than for the 2,000-mL transfusion and still maintain the same total leachable concentration for a patient.
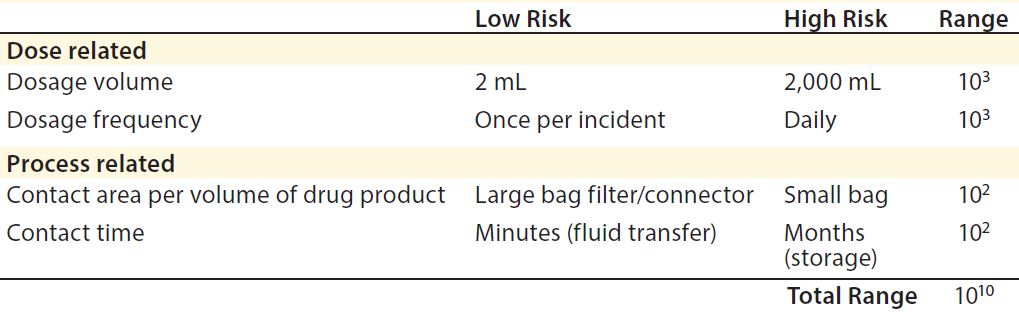
Table 2: Safety concerns for leachables in final products (3)
Overall, the impact of both process-related and dose-related factors is cumulative. They can vary the acceptable leachable concentration per volume (e.g., allowable leachable per milliliter) by many orders of magnitude (3) (Table 2). Considering those factors is critical when establishing required testing and allowable limits for leachables and extractables in a biomanufacturing process.
Comparing Cellular iTx and MAb Bioprocessing Strategies
Protein-based biotherapeutics are more challenging to manufacture than small molecules because biologics are more likely to be affected chemically by leachables and environmental changes. Challenges for therapeutic cells are likely to be even more profound.
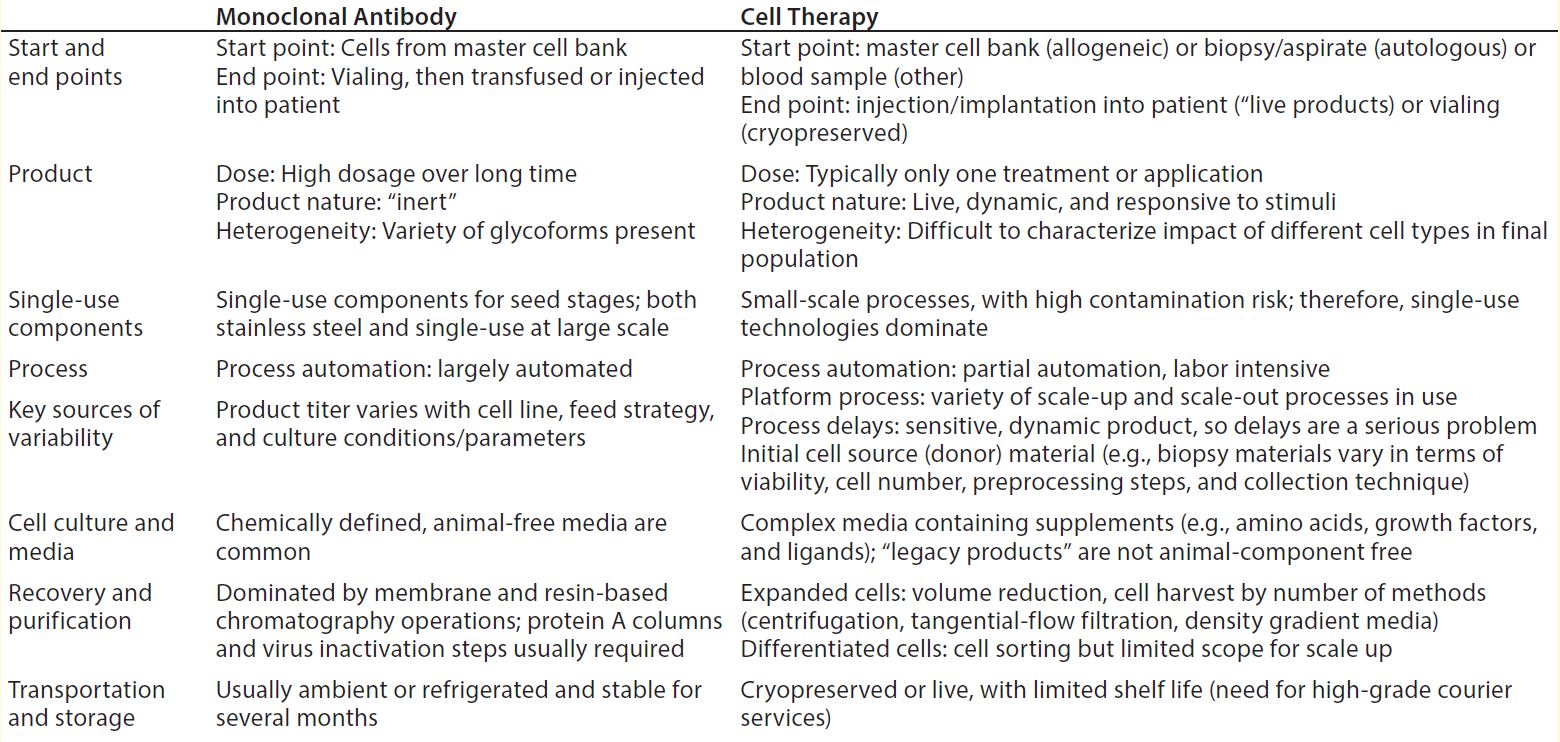
Table 3: Comparing MAbs and therapeutic cell products
Commercial production cell lines for biologics (e.g., CHO, HEK293, and NS0) are engineered to exhibit robustness and stability throughout a biomanufacturing process (4). So it is clear that therapeutic cell lines will have further heightened sensitivity to environmental conditions because of a number of differences between MAbs and cell therapies (Table 3) (4). iTx cell lines display complex nutritional demands depending on exogenous growth factors, responsiveness to endogenously produced growth factors, heightened response to production parameters (e.g., pH and dissolved oxygen), and responsiveness to mechanical forces (4). As such, iTx autologous therapies will most likely be more susceptible than other therapies to influences from leachables and extractables present throughout manufacturing processes.
Development of cell-based iTx requires repurposing existing single-use biomanufacturing platforms (4). That task will be the industry’s primary focus because many current cellular iTx clinical trials are investigating autologous therapies, whereas allogeneic immunotherapies are handled similarly to commercial biotherapeutics. Autologous iTx are generally cultured in much smaller volumes (1–30 L), and they therefore have a greater ratio of culture bag to total volume, which increases the amount of contact that cells have with single-use technologies (e.g., connecting tubing and the polymer films used in the rocking-motion culture bags). Source material variation (due to differing acquisition protocols and indivitual patient cell make up) are unpreventable, but control of attributes such as leachables and extractables can help reduce variability during manufacturing.
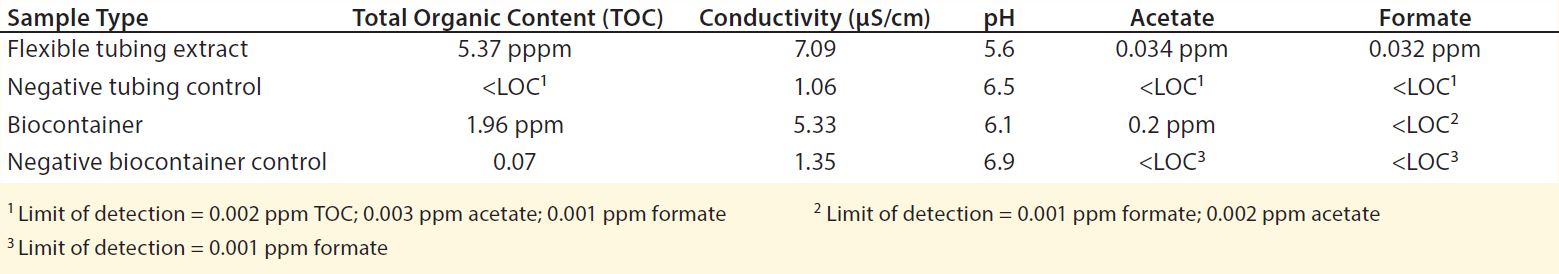
Table 4: Water extract values from flexible tubing and biocontainers (12)
To evaluate potential leachable and extractable accumulations in iTx processes, manufacturers must conduct in-house testing procedures for tubing and biocontainers used. For example, sections of flexible tubing can be cut into strips and filled with a model solvent or water. Once each end is sealed, the tube sets incubate for 72 h at 21 °C in a shaken incubator (11). Biocontainers can follow a similar procedure, but with the incubation period set at 30 days (or longer) and at 40 °C to accommodate for longer contact times and incubation temperatures that are common in tube sets during cell-expansion phases of manufacture (11). Table 4 shows example data produced by a study of this type.
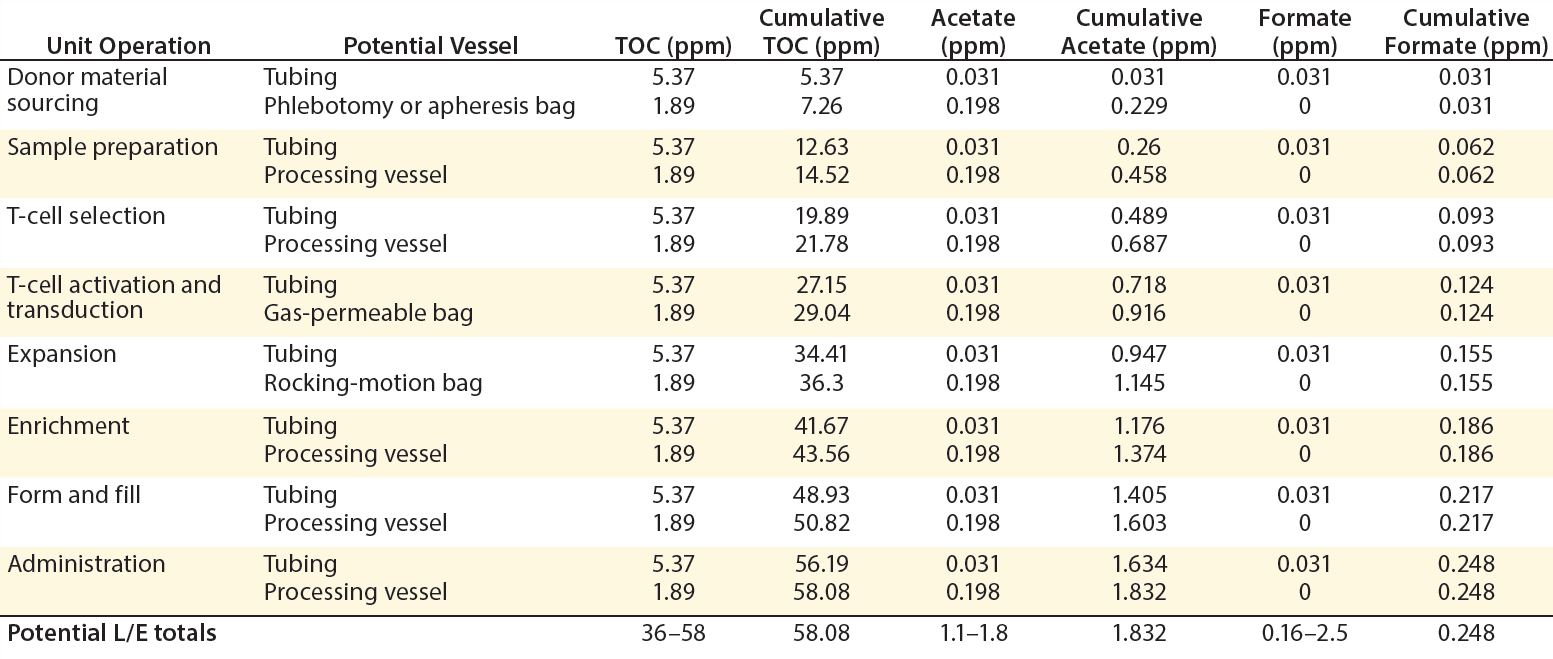
Table 5: Estimated leachable and extractable levels in immunotherapy biomanufacturing
Complementing the complexity of autologous manufacturing processes, the high number of processing steps as well as product contact with different single-use systems raises concerns about leachables and extractables. Tubing and biocontainer values from a previous study (Table 4) formed the basis of risk-indexed estimates for potential leachable and extractable amounts that could possibly be achieved during a “typical” autologous cellular immunotherapy manufacturing process as laid out in Table 5 (13). To adjust for risk, these calculations took into account the duration of contact for each unit operation.
Table 5 shows the potential for extractables and leachables to be introduced at each stage of an iTx manufacturing process (vector generation is omitted from Table 5). However, in the absence of well-characterized or platform iTx manufacturing processes, it is difficult to determine a uniformly acceptable extractable and leachable profile within an SOP. We conducted a Pareto analysis to evaluate and identify points of greatest bioprocess risk arising from extractables and leachables.
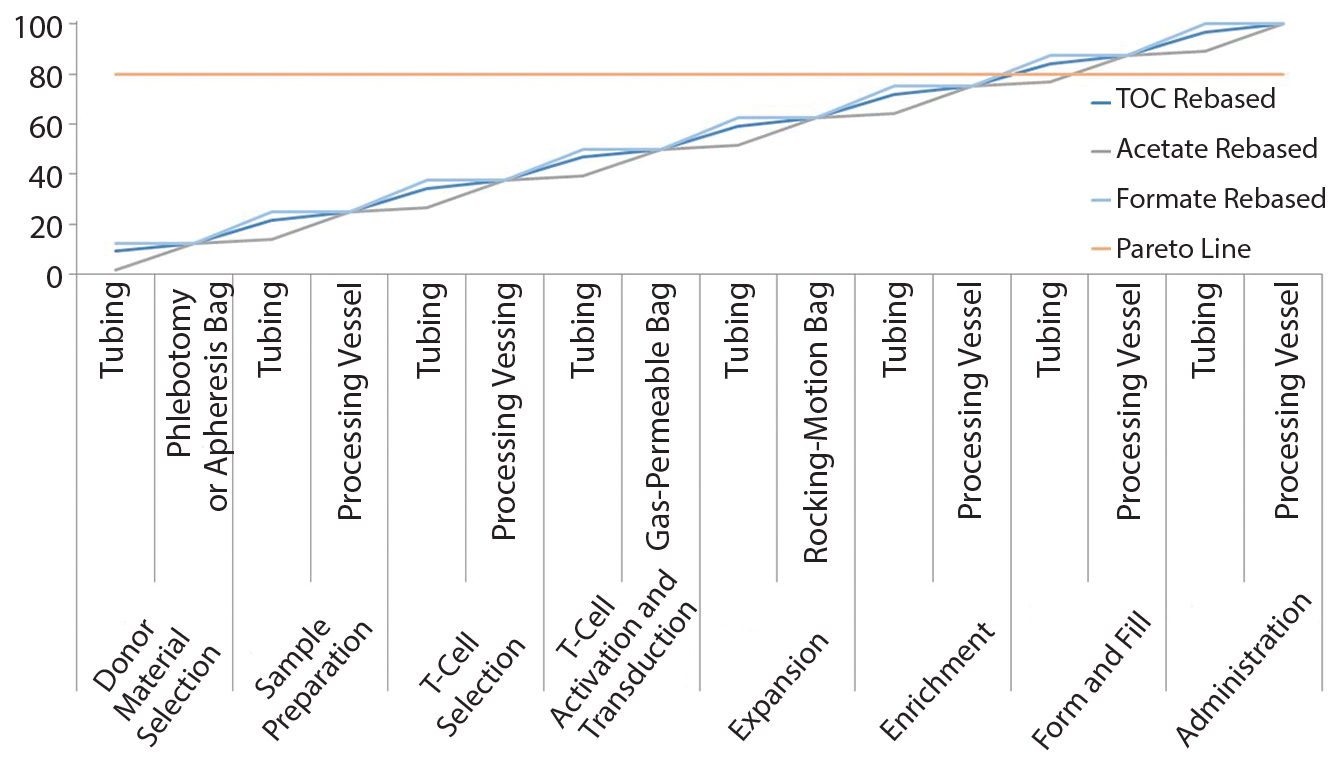
Figure 2: Pareto analysis of bioaccumulation of leachables and extractables in iTx manufacturing assuming linear accumulation throughout the bioprocess
Figure 2 shows estimated bioaccumulation rates for extractables and leachables in an exemplar iTx manufacturing process (assuming a linear accumulation profile). Process fluids — including therapeutic cells — have an equal bioaccumulation risk throughout all unit operations. This analysis suggests that the 80% of potential extractables and leachables are accumulated at the interface of enrichment and formulation and fill stages.
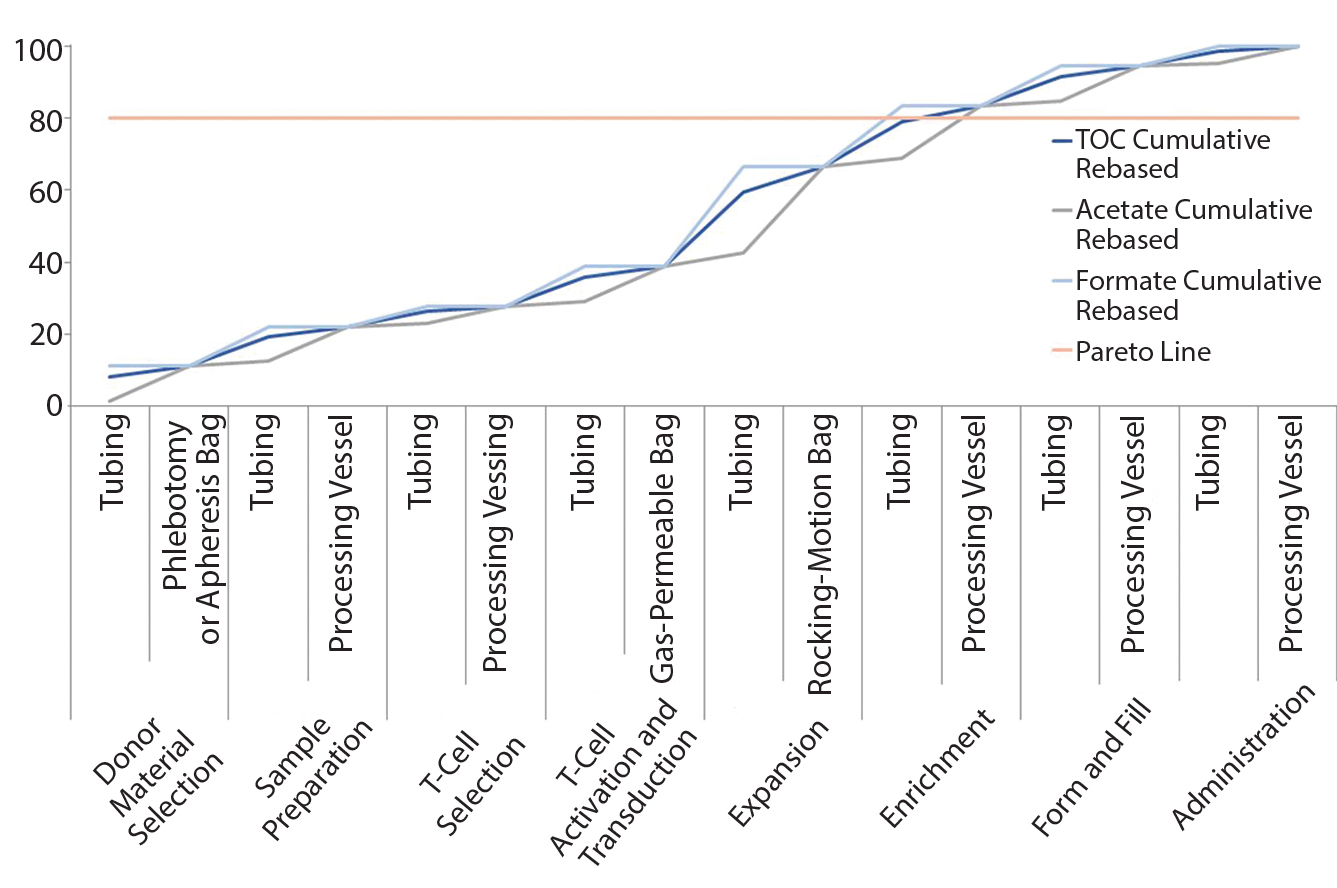
Figure 3: Pareto analysis of bioaccumulation of leachables and extractables in iTx manufacturing assuming nonlinear accumulation throughout the bioprocess
Figure 3 improves upon Figure 2 by using a relative-risk index (Table 6) to estimate the relative potential exposure of process fluids to extractables and leachables throughout a bioprocess. For example, the greatest contact with single-use components (in terms of both time and surface area exposure per unit cell) is likely to be during the cell-expansion stage.
Comparing Figure 2 with Figure 3 clearly shows that introducing a relative-risk index changes thecurve shape closer to a classical S-curve and shifts it modestly toward the y-axis. Thus, the critical 80% limit is reached earlier in a bioprocess. However, further validation of estimated relative-risk index parameters is required to validate and optimize this finding.
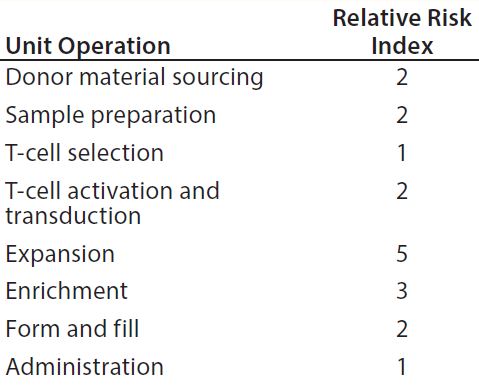
Table 6: Estimating the relative potential exposure of process fluids to extractables and leachables throughout a bioprocess
Steps to Mitigate Leachables and Extractables
The first step to mitigating risk is a comprehensive risk assessment strategy for determining an immunotherapy’s critical quality attributes (CQAs) and how they are influenced by the presence of unwanted contaminants in the form of leachables and extractables. Identifying specific leachables and extractables and then quantifying allowable concentrations per dose down to parts-per-billion levels (11) is required to detect the trace amount of contaminants present on all films (14). Manufacturers can help reduce the burden of on-site testing for leachables and extractables by working with suppliers of various tubing, connectors, filters, media bags, rocking-motion bags, and other storage bags. Such suppliers can provide validation criteria for their single-use components. Investigating the availability of films that are specifically formulated and designed to exhibit very low leachable and extractable profiles also can help manufacturers prevent regulatory delays in the product commercialization phase (7).
An example of such testing performed by one major supplier of such single-use systems includes the following release criteria in regard to internal testing of a film exhibiting low leachables and extractables:
- USP Class VI Plastic Test for Biological Safety (intracutaneous test, systematic injection test, and implantation test) compliant with 21 CFR regulations
- extractables determined through various analytical tests using pure solvents such as reverse osmosis water, ethanol under defined extraction conditions (duration, temperature, shaking or flowing, volume, and so on). Extracts analyses generally involve determination of the amount of nonvolatile residue (NVR) and the total oxidizable content (TOC), pH, and conductivity.

Figure 4: Single-use bag supplier lot-release testing, analytical scheme for extractables analysis (used with permission, reference 16)
Lot-release tests on bags involve a wide range of analytical tools such as gas chromatography–mass spectrometry (GC-MS), high-performance liquid chromatography (HPLC), or Fourier transfer infrared (FT-IR) spectroscopic analysis, liquid chromatography–MS (LC-MS), and others. Manufacturer-supplied data can help reduce the burden of on-site analytical, quality control (QC), and quality assurance (QA) groups. Some suppliers offer individual batch-release records with batch-specific, lot-release testing data. And some suppliers provide one film for use in all unit operations of immunotherapy manufacture, which clearly reduces qualification and validation steps for that film or process.
Using these tests and techniques — along with those for tubing and other biocontainers — manufacturers can develop a quantitative and qualitative extractables profile for each component used in a manufacturing process. For each component, such data will reduce or remove the need for further testing of a single-use system as a whole, mitigate risks, and ultimately, protect patients (11).
Collaborative Efforts Are Needed
The personalized nature of autologous cellular immunotherapies necessitates implementation of single-use technologies for commercial manufacture. Therefore, developing a robust and comprehensive risk analysis methodology to identify and quantify leachables and extractables from single-use components is critical to manufacturing success. The contaminants discussed here have been shown to affect product quality, production consistency, and stability of established therapeutic products such as MAbs and recombinant proteins. The increased sensitivity of cell lines commonly used for cellular immunotherapy products make the risks associated with leachables and extractables even more critical. Establishing close relationships between cell therapy manufacturers and suppliers of single-use technologies can be beneficial for both parties. Low-leachable and -extractable films can help manufacturers continuously improve the robustness of their single-use manufacturing processes. On-going communication between manufacturers and suppliers can help establish new release criteria for both single-use technologies and for final drug-product release testing. Manufacturers working together with regulators to establish release criteria will provide a more clearly defined path forward, especially when such criteria take into account the impact of leachables and extractables on the developing and expanding field of cellular immunotherapies.
Acknowledgments
We express our sincere thanks to the following organizations that have contributed to the CASMI Translational Stem Cell Consortium (CTSCC) as funding and events partners, without whom the consortium and the benefits it will bring to stem cell translation would be constrained: GE Healthcare, CCRM, Sartorius Stedim Biotech (formerly TAP Biosystems), Lonza, CIRM, SENS Research Foundation, UK Cell Therapy Catapult, NIH Centre for Regenerative Medicine, NYSCF, ThermoFisher Scientific, Eisai, Medipost (US), Medipost (Korea), Celgene, Roche and Oxford Biomedica. We also sincerely thank Christopher A. Bravery, principal consultant of Consulting for Advanced Biologics, for his insights on the regulatory landscape associated with leachables and extractables. D.A.B. gratefully acknowledges personal funding from the Oxford Musculoskeletal NIHR BRU, the Said Foundation and the SENS Research Foundation. J.A.S gratefully acknowledges support from the CASMI Translational Stem Cell Consortium (CTSCC).
Conflict of Interest Statement
The content outlined herein represents the individual opinions of the authors and may not necessarily represent the viewpoints of their employers. D.A.B. is a stockholder in Translation Ventures Ltd. (Charlbury, Oxfordshire, UK) and IP Asset Ventures Ltd. (Oxford, Oxfordshire, UK), companies that among other services provide cell therapy biomanufacturing, regulatory, and financial advice to pharmaceutical clients. J.A.S is a consultant of IP Asset Ventures Ltd. D.A.B. is subject to the CFA Institute’s codes, standards, and guidelines, and as such, this author must stress that this piece is provided for academic interest only and must not be construed in any way as an investment recommendation. Additionally, at time of publication, D.A.B. and the organizations with which he is affiliated, may or may not have agreed and/or pending funding commitments from the organizations named herein.
References
1 Clarke D. Single-Use Technologies in Cell Therapy. BioProcess Int. 11(3) 2013: S12–S17.
2 Mire-Sluis A, et al. Extractables and Leachables: Challenges and Strategies in Biopharmaceutical Development. BioProcess Int. 9(2) 2011: 14–23.
3 Bestwick D, Colton R. Extractables and Leachables from Single-Use Disposables. Bioprocess Valid. 7(3) 2010: S88–S94.
4 Brindley D, et al. Cell Therapy Bioprocessing Technologies and Indicators of Technological Convergence: A Concise Review. BioProcess Int. 12(3) 2014: S14–S21.
5 Good Manufacturing Practices, Medicinal Products for Human and Veterinary Use. European Commision: Brussels, Blegium, 1998.
6 Equipment Construction. Code of Federal Regulations, Food and Drugs. US Goverment Printing Office: Washington DC, Revised April 2006.
7 Kauffman J. Identification and Risk Assessment of Extractables and Leachables. Pharma. Technol. February 2006.
8 Whitford W. Single-Use Systems As Principal Components in Bioproduction. BioProcess Int. 8(11) 2010: S34–42.
9 BPSA Board of Directors. Advancing Single-Use Worldwide. Mission Statement.
10 Eibl D, Eibl R, Kohler P. Single Use Technology in BioPharmaceuctical Production. Report of the Temporary Working Group, DECHEMA: Frankfurt, Germany, March.
11 Ding W, Martin J. Implementation of Single-Use Technology in Biopharmaceutical Manufacturing: An Approach to Extractables/Leachables Studies, Part Two: Tubing and Biocontainers. BioProcess Int. 7(5) 2009: 46–51.
12 Hammond M, et al. A Cytotoxic Leachable Compound from Single-Use Bioprocess Equipment That Causes Poor Cell Growth Performance. Biotechnol. Prog. 30(2) 2014: 332–327.
13 Brindley D, et al. The Potential Application of Real-Time Release Testing for the Biomanufacture of Autologous CellBased Immunotherapies. BioProcess Int. 13(4) 2015: S34–S43.
14 FlexSafe RM Extractables Analysis. Sartorius Stedim Biotech GmbH: Goettingen, Germany, 2014. 15 Minow B, Rogge P, Thompson K. Implementing a Fully Disposable MAb Manufacturing Facility. BioProcess Int. 10(6) 2012: 48–58.
Corresponding author Kevin A. Lannon is the North American field marketing manager for regenerative medicine at Sartorius Stedim Biotech; kevin.lannon@ sartorius-stedim.com. James A. Smith is a research associate at the Oxford-UCL Centre for Advancement of Sustainable Innovation (CASMI) Translational Stem Cell Consortium (CSTCC) and a DPhil candidate at the University of Oxford’s Nuffield Department of Orthopedics, Rheumatology and Musculoskeletal Sciences (NDORMS). Kim Bure is director of regenerative medicine at Sartorius Stedim Biotech. Corresponding author David A. Brindley is the CookseyBotnar-Saïd Fellow in Healthcare Translation at the University of Oxford’s Nuffield Department of Orthopedics, Rheumatology and Musculoskeletal Sciences (NDORMS) and the Saïd Business School; research fellow at the Harvard Stem Cell Institute; senior research fellow at the University College London Centre for Behavioral Medicine; and Member of the Stanford-UCSF FDA Centre for Regulatory Science and Innovation (CERSI); david.brindley@sbs. ox.ac.uk and david.brindley@ndorms. ox.ac.uk.