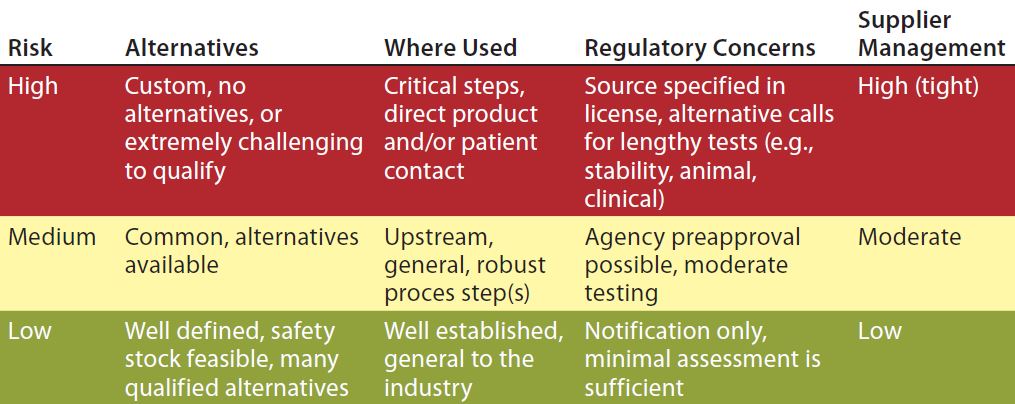
Table 1: Risk-level matrix
Ensuring a continuous supply of safe medicines is a key objective for the pharmaceutical industry and health authorities alike. A critical component to that end is maintaining a reliable supply of qualified raw materials (RMs) used in drug production. However, changes in suppliers, their processes, their providers, and consequently the materials they supply can occur (for a number of reasons) at any time during the life cycle of drug production. A product-supply organization therefore must be prepared to address such changes in time to prevent interruption of patients’ supply.
The biomanufacturing industry recognizes that it needs to apply a risk-based RM control strategy in line with pertinent regulatory and other guidelines based on an understanding of possible effects on product quality (1). Here we present a risk-based, bestpractice approach and case study for mitigating the risk of drug-product supply shortages based on the availability of qualified raw materials.
Our Approach
In biomanufacturing, RMs include everything in the bill of materials (BoM). Examples include chemicals (such as those found in media, buffers, resins, excipients, process agents, and cleaning solutions), disposable systems, and packaging. These RMs are procured from a broad range of different suppliers (2). Users and suppliers both want to maintain their business relationships, but risks include financial stability, supply, performance, capacity, and quality issues.
To best mitigate a risk, it must be well defined. Level of risk can be determined according to a number of criteria. Generally it is higher when RMs are less common or when qualifying alternatives is more challenging, when they are used in more critical process steps, when regulatory concerns around them are high, and/or when the need for tight supplier management is great (Table 1). However, decisions regarding which materials to address first, which tools to use, and how to order them in priority (see “Toolbox” box)
| Toolbox |
| Contracts Strong relationship with supplier Lead time Second site Safety stock Second source qualification |
take additional considerations into account. These center on the best application of available resources (triage methodology, Table 2). Because demand and supply are dynamic, their urgency levels are continuously monitored and (re)classified (tracking methodology, Table 3), making it possible for materials to shift in relative urgency (among A, B, and C clusters in Table 2).
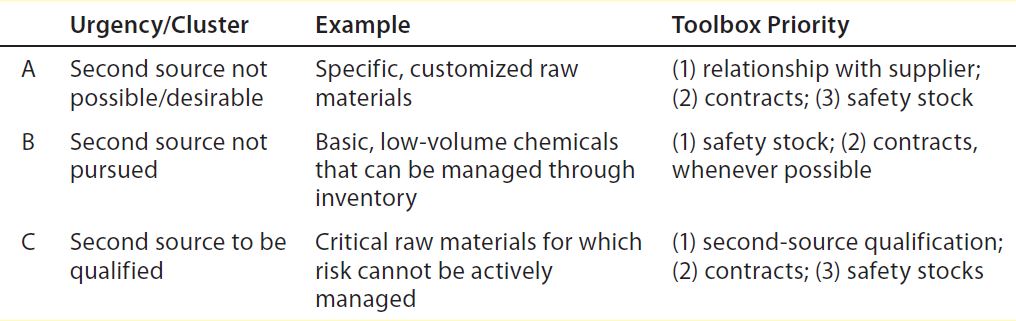
Table 2: Triage methodology; materials clustered based on decision whether to second-source
Keep in mind that risk-management tools inherently differ, and the level of control a sponsor company has over them can change continually. So they have to be assessed continuously for effectiveness and priority. Contracts, purchase orders, quality agreements, and user requirements all are subject to a two-way agreement between business parties. Expecting a supplier to agree to high-liability language when spending is low can be a challenge.
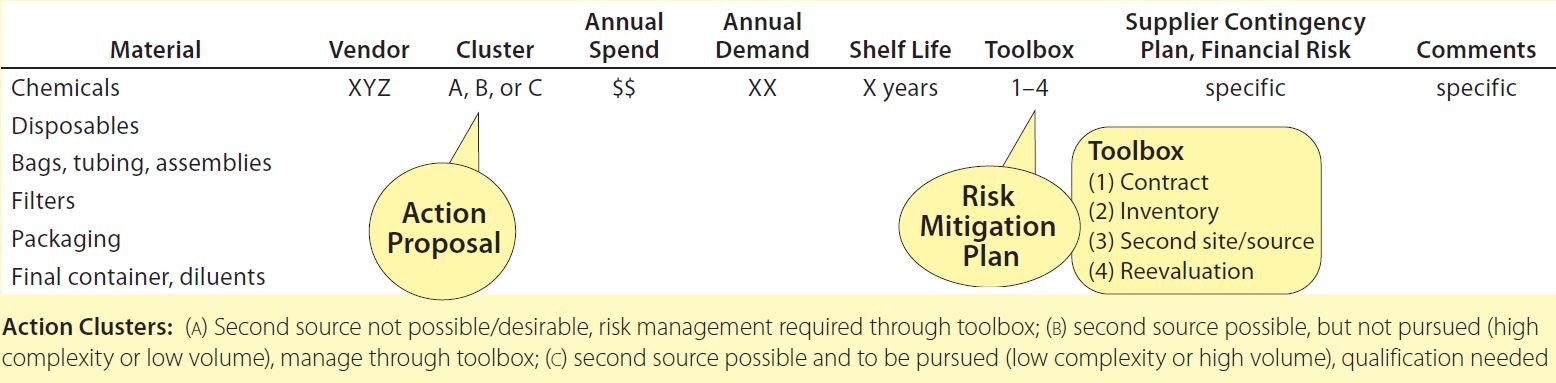
Table 3: Tracking methodology for example material types
The importance of maintaining a strong relationship with suppliers cannot be understated, and efforts to that end can pay high dividends for both parties. Short lead times are an obvious business and supply advantage, yet concerns about events that can’t be controlled by either partner (e.g., natural disasters) may make it prudent to consider a second material source. Safety stocking is not best or possible for all RMs. For example, physical space constraints and/or working-capital burdens may preclude keeping a stock in house. In a few instances, such considerations make it appropriate instead to qualify a second-source supplier as the top priority (Table 2); in other instances, other tools are prioritized (Table 3).
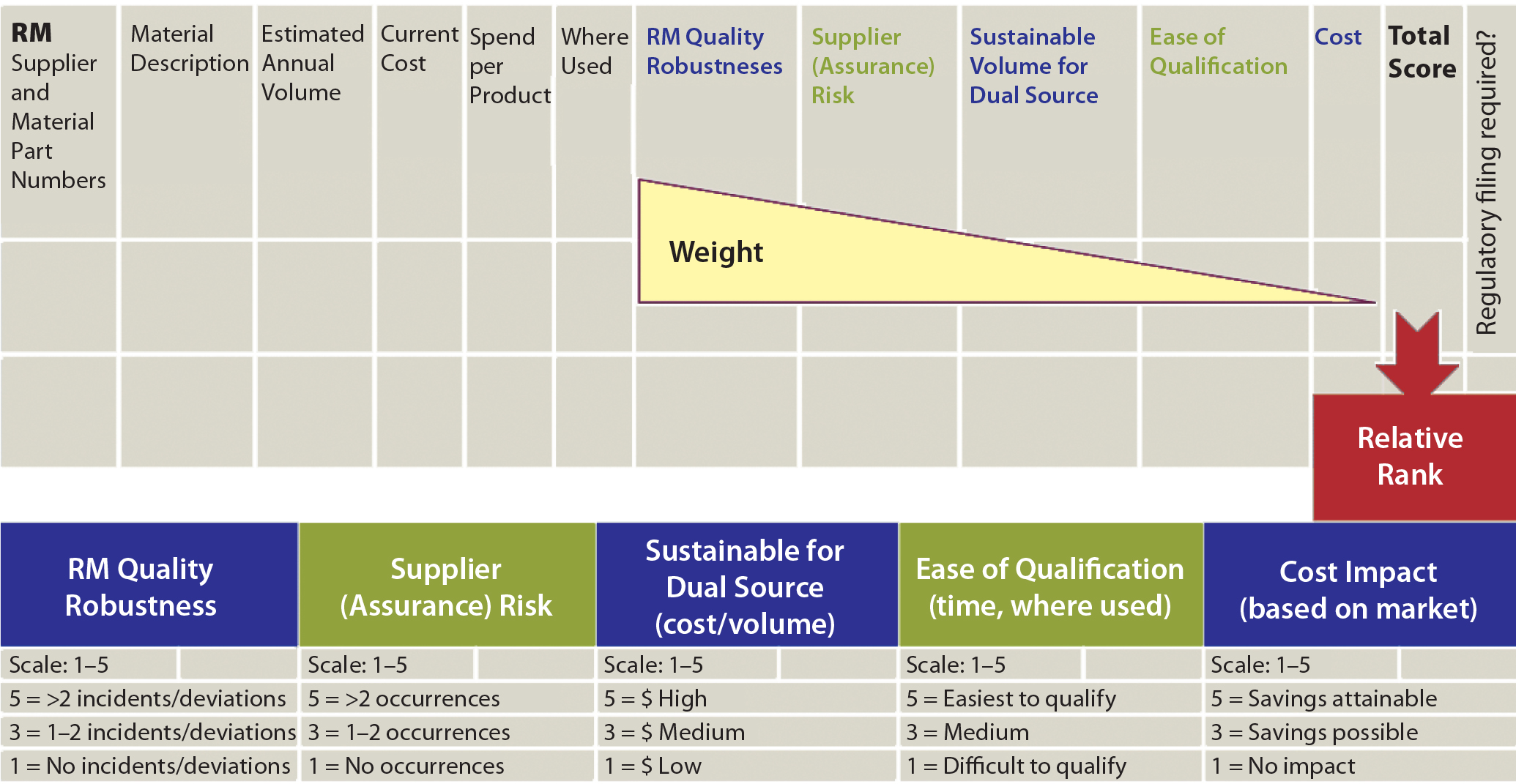
Figure 1: Dual-sourcing decision matrix
When second-source qualification is determined to be best for multiple RMs (but qualifying them all at once is infeasible), using a prioritization decision matrix is helpful. A weighted scoring system is based on five criteria in decreasing weight: quality risk (can be determined based on historical incidents), supply assurance risk, sustainable volume for dual source, ease of qualification, and cost (Figure 1). This exercise produces a prioritized list of RMs for which a second source is to be qualified. Such a qualification program is described below.
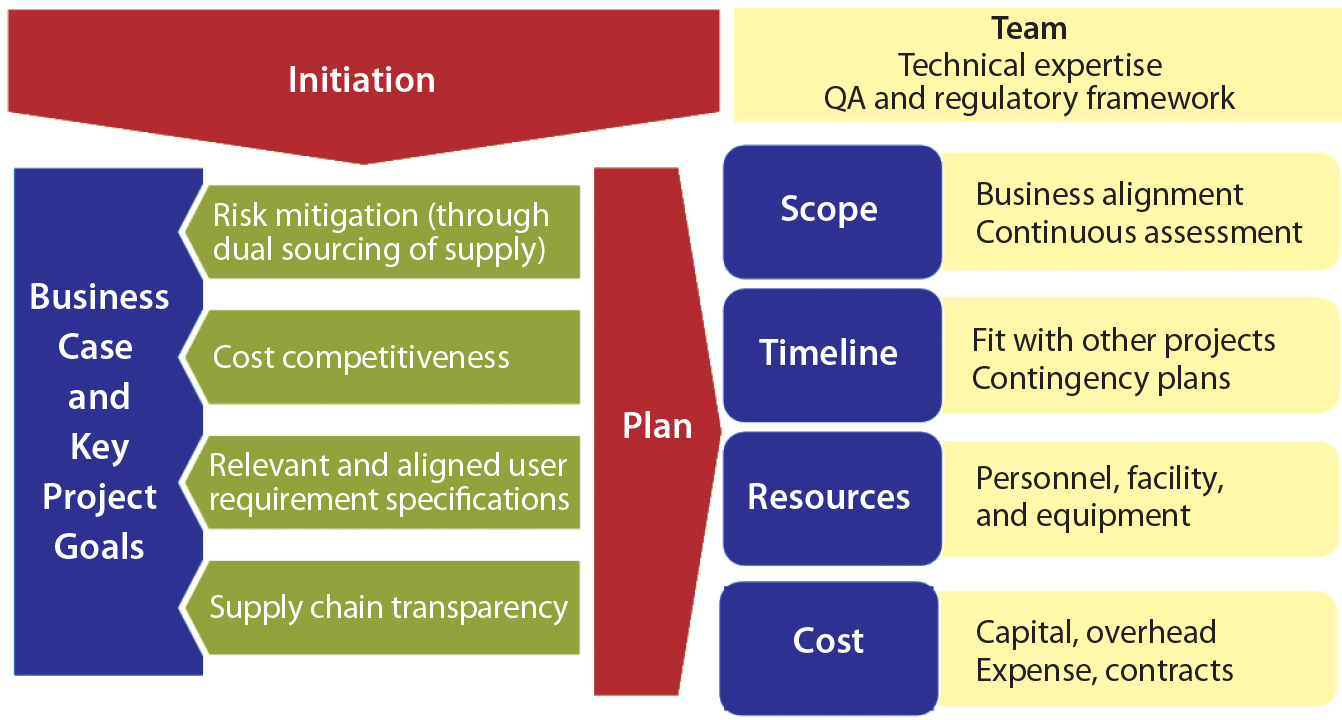
Figure 2: Case study: cell culture dry powder media supplier and material qualification
Case Study
Supplier qualification for dry-powder cell culture media can serve as a representative case study because the challenges involved are common among companies throughout the biomanufacturing industry. A qualification plan is put into place with appropriate justification, highlighting goals and potential benefits (business case and key project goals) and defining the project framework (scope, timeline, resources, and cost; Figure 2).
Execution toward implementation can then follow, being driven by a strategy that in turn considers business and compliance requirements. These requirements could include attributes such as quality, innovation, cost, service, and assurance of supply. Ideally, several suppliers would meet those requirements (which can be determined through review of their proposals and responses to a questionnaire). They would be invited then for a day of face-to-face presentations and discussions in which the biomanufacturer presents pertinent elements of its current business, future prospects, and expectations from partnerships with its suppliers. Such a “supplier day” can greatly help differentiate and facilitate selection of the most suitable supplier to pursue based on mutual fit and best chances for mutual success.
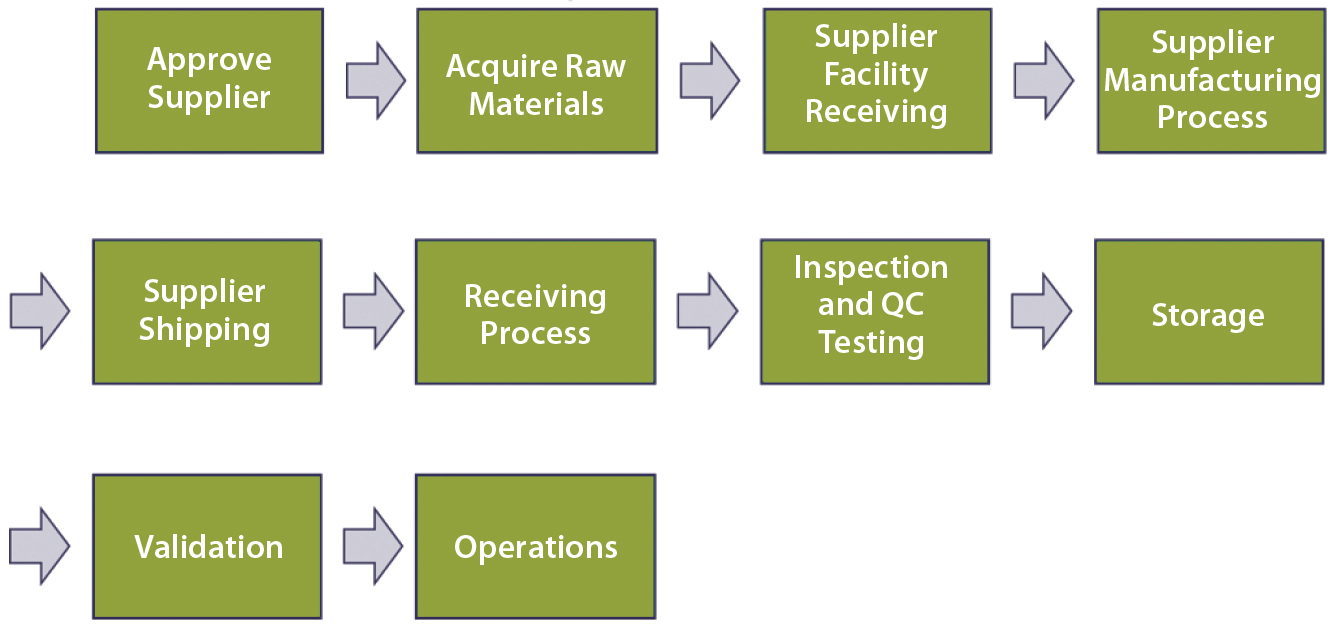
Figure 3: Top–down, end-to-end process map
Emphasis would then shift toward technical qualification (of the supplier and RM), the scope of which would be determined by risk level (the higher the risk, the broader the scope, as explained below). The level of risk can be quantified using operational excellence tools such as failure modes and effects analysis (FMEA). Performing a comprehensive FMEA would require a detailed process map developed (by a team of subject-matter experts, SMEs) to the level of independent operational steps. Figure 3 is an example of high-level, top-down end-to-end mapping.
Each top-level process step should be further broken into subprocesses, with those further broken as appropriate (not shown). End-to-end indicates a starting point at the vendor’s own supply chain. In the dry-media example, that would be each individual component to be processed (blended and milled) in the supplier’s manufacturing process: salts, sugars, amino acids, vitamins, trace metals, lipids, and so on. That process would end when the RM is in the form in which it will be used (buffered liquid media to be fed to cells in production culture).
Once a process has been mapped, the FMEA process begins. As a prevention tool for identifying potential risk, FMEA also can be used to help eliminate failures, problems, and/or errors in processes before they cause undesirable effects. The FMEA exercise starts with ground rules. Setting them includes scaling for three factors: severity, occurrence, and detection.
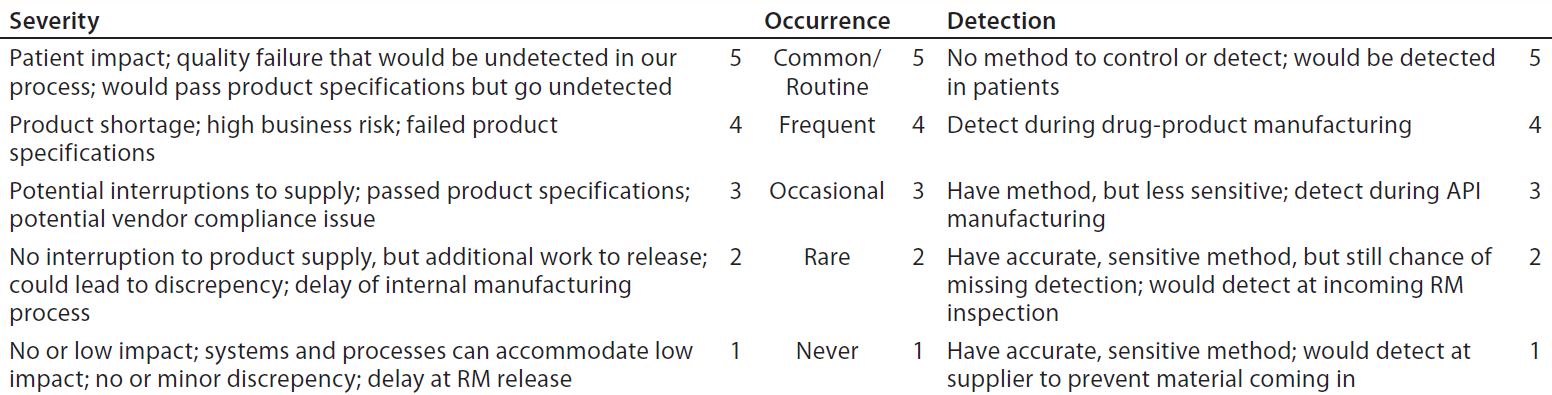
Table 4: FMEA scales setting (RM = raw materials)
Table 4 shows scales tailored to RM supply failure modes and effects. A risk-analysis team next runs through the FMEA ranking process outlined below. Note that one potential failure mode can have multiple effects and/or causes. In such cases, a risk priority number (RPN) would be calculated for each independently by multiplying the three factors, S × O × D (see below).
An FMEA exercise typically includes the following steps:
- Brainstorm potential failure modes (for each of the process steps).
- List potential effects of each failure mode.
- Assign severity (S) ratings to each potential effect.
- Brainstorm potential causes of each failure mode.
- Assigning occurrence (O) likelihood rating for each potential cause.
- Examine current controls or detection methods for each potential cause.
- Assign detection (D) or control rating for each failure cause.
- Calculate the RPNs.
- Propose and follow-up on corrective actions to minimize risks and reevaluate RPNs.
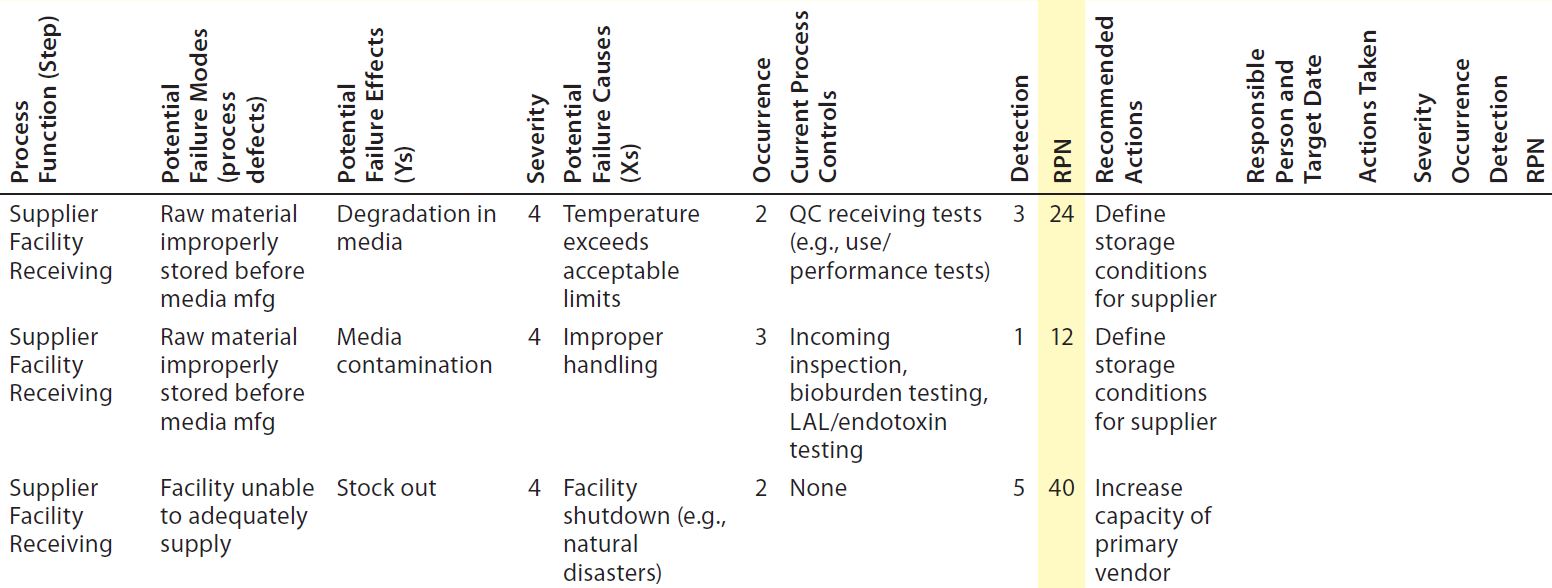
Table 5: Example FMEA worksheet; Severity (S) × Occurrence (O) × Detection (D) = Risk Priority Number (RPN = S × O × D)
FMEA can take several meetings to complete, so a consistent composition of SME team members (if feasible) greatly facilitates the process because everybody is familiar with the “ground rules” for ranking throughout. Some teams find it more effective to prepopulate (at least portions of) the FMEA chart in advance and then simply review them in a team setting. Table 5 is an example of a single process step.
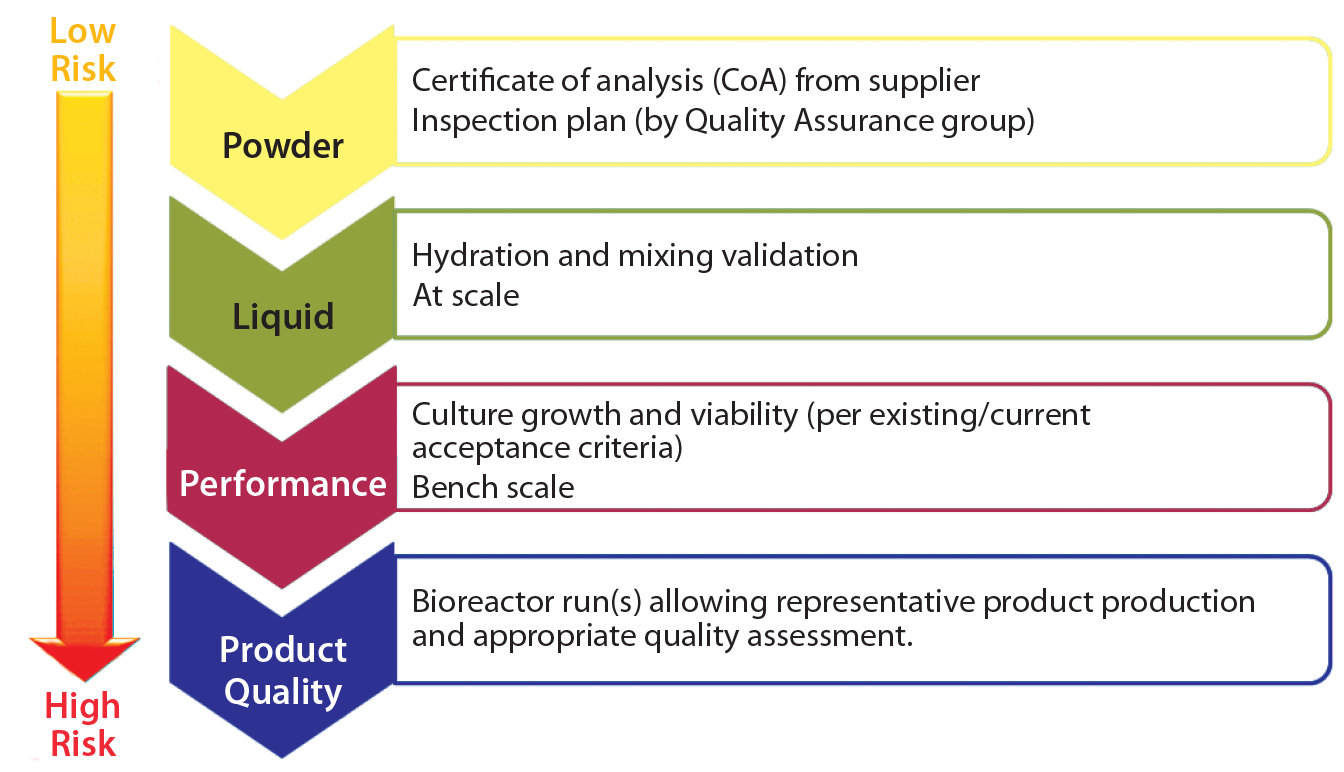
Figure 4: Qualification plan per level of risk
Following the FMEA exercise, qualification plan details can be developed based on the overall level of risk determined after corrective actions have been applied. Generally, higher risk requires a qualification plan of broader scope (Figure 4). This could entail:
- RM testing (e.g., dry medium powder) as reported on the supplier’s certificate of analysis (CoA) and in-house material inspection and testing
- testing performed on the form in which the RM is used (e.g., following hydration, including at-scale mixing validation)
- process performance determined through functional testing (e.g., by testing cell growth and viability)
- release assays based on release specifications and/or acceptance criteria for the full cohort of product quality attributes (PQAs).
The latter includes structural, functional, and impurities testing (and possibly extended characterization tests). Because dry medium powder is used in upstream operations (cell culture), an FMEA team also would need to assess its potential impact on downstream operations (e.g., isolation and purification).
In some cases, at-scale validation in commercial production presents significant challenges that could limit the number of runs that would be feasible to perform. So we developed an approach to qualification of smallscale bioreactors for use in validation and have demonstrated its application (3, 4). Such an approach could reduce risk upon implementation because using qualified small-scale systems provides flexibility that enables a larger number of validation runs to be performed than with a large “at-scale” platform. Note that this is in line with current regulatory-agency expectations for a science-based approach to process validation.
The timeline from (second RM) supplier qualification project initiation to implementation will be unique to each company and even to each specific platform within a given company. Generally, it could take one to three years to complete. This entails a vendor survey and selection process; agreements, audits, and compliance assurance; and tests and validations of RMs, processes, and products at different scales, as appropriate.
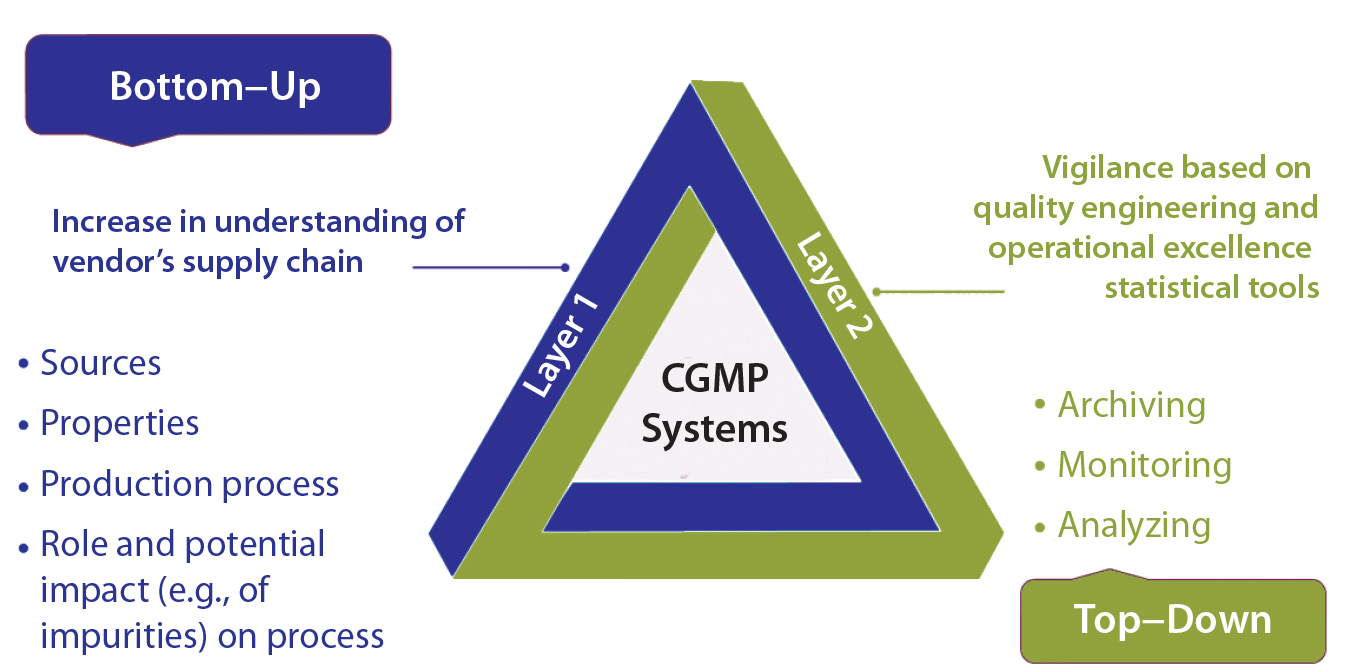
Figure 5: Postimplementation tracking
Once a new supplier’s material has been implemented, continuous tracking will be important as experience accrues (Figure 5). This can be accomplished at complementary levels: from the bottom up for technical expertise (both the user’s and the supplier’s) regarding a vendor’s own RM supply chain; and from the top down for quality engineering and operational excellence statistical tools to archive, monitor, and analyze relevant data for vigilance.
Raw Materials Risk Mitigation
We have presented herein an approach to determine RM risk and urgency levels and prioritize risk-mitigation tools accordingly. For the option of second-supplier sourcing, we recommend a layered approach. Our case study describes the qualification of a second source and/or supplier for dry media powder RM.
References
1 Kolhe P. Raw Material Control Strategy Key to Overall Control. PDA Letter L1(1) 2015: 32–33.
2 Bowman D. A Quick Guide for Sourcing Biopharmaceutical Raw Materials. BioProcess Int. 13(3) 2015: 51–52.
3 Shimoni Y, et al. Qualification of ScaleDown Bioreactors: Validation of Process Changes in Commercial Production of Animal-Cell–Derived Products, Part 1 — Concept. BioProcess Int. 12(5) 2014: 38–45.
4 Shimoni Y, et al. Qualification of ScaleDown Bioreactors: Validation of Process Changes in Commercial Production of AnimalCell–Derived Products, Part 2 — Application. BioProcess Int. 12(6) 2014: 54–61.
Acknowledgments
For their contributions to developing the approach presented herein, we thank these colleagues: Holly Baraona, Russell Wong, Matt Muldoon, Shelly Bustillos, Dale Dwier, Tobias Pace, Ruben Omega, and Yongchun Zhang. We are also grateful to Olivia Krampe, Jay Benson, and Joerg Heidrich for their sponsorship and encouragement.
Yuval Shimoni is principal engineer, and Venkatesh Srinivasan is director of manufacturing sciences; and Markus von Gruchalla-Wesierski is director and head of west-coast procurement at Bayer HealthCare LLC, 800 Dwight Way, Berkeley, CA; 1-510-705-5775; yuval.shimoni@bayer. com, www.bayerhealthcare.com. This work was presented by Shimoni at WTG Events’ February 2015 BioManufacturing Summit in Orange County, CA, as part of a discussion on approaches to ensuring continuous supply of raw materials needed for drug production.