
PALL LIFE SCIENCES (WWW.PALL.COM)
Monoclonal antibodies (MAbs) serve important medical needs in cancer treatment as well as that of autoimmune and infectious diseases (1). Antibodies are also widely used in clinical diagnostic assays. They can be coated on solid surfaces to bind specific analytes, conjugated to reporter molecules (either as whole antibodies or fragments) for analyte detection, used in sensitivity panels for lot-release testing, and supplied as positive controls in diagnostic kits (2). Our study evaluates the use of hydrophobic charge-induction chromatography (HCIC) for purification of MAbs that are of interest for diagnostic applications. Our objective was to obtain ≥90% purity with ≥90% IgG recovery from a single chromatographic step — a challenging objective with most experiments conducted using feedstock derived from protein-supplemented growth media. To assess the versatility of the HCIC approach, we used both concentrated and unconcentrated dilute feedstocks as well as a number of different antibody isotypes and source species. We also isolated MAbs from different growth media formulations.
Purification platforms for diagnostic antibodies can involve a single chromatographic step, as in the work reported here. But our findings also should interest process engineers developing multistep purification schemes. HCIC has been evaluated in development of multistep schemes for purification of therapeutic antibodies using strategies based on protein A affinity capture and those that do not rely on it (3–5).
In diagnostic and research applications, MAbs of different immunoglobulin classes and isotypes are derived from a broad range of species sources. Veterinary therapeutic applications can require antibodies from a number of species sources. Such antibodies are often poorly retained on protein A sorbents (6). Purification of murine IgG1 on protein A sorbents, for example, typically requires binding at pH ≥8.5 and then augmenting feedstock with concentrations of binding-promoting salt that approach the antibodies’ precipitation point (6).
By contrast, no enhanced binding conditions are required when using the HCIC sorbent we chose (7). Murine IgG1 and IgG2a will bind to it directly from clarified cell-culture supernatant or from dilute buffer at pH ≤8.0 (7, 8). Studies have shown similar binding capacity values for murine IgG1 and IgG2a during HCIC: 37 and 34 mg/ mL, respectively (9, 10). Immunoglobulin E, which is poorly retained on protein A, also has been purified by HCIC (11, 12). Brief reports have also described purification of IgM with HCIC (10, 13). To explore its broad selectivity further, we include purification of murine IgG1, murine IgG2a, and ovine (sheep) IgG1.
Broadly speaking, process developers are increasingly focused on strategies to support enhanced process economics and robustness. In that context, sorbents carrying synthetic chemical ligands are inherently less costly than those based on protein ligands. Moreover, the former are typically compatible with stringent cleaning agents (e.g., 1 M sodium hydroxide) than the latter. These considerations suggest a potentially expanded role for HCIC in purification platform development.
Affinity chromatography on protein A sorbents remains the most widely used strategy for MAb capture and initial purification (14–17). But a number of researchers have investigated strategies that do not rely on protein A (3, 18), or that make use of sorbents carrying immunoglobulin-selective, mimetic ligands (19, 20). Development of platforms without protein A has been facilitated by a considerable body of literature describing use of other chromatographic modes of in conjunction with protein A affinity chromatography. Some such techniques also have been evaluated as alternatives to protein A for capture chromatography: cation-exchange chromatography (CIEX) (21), hydrophobic-interaction chromatography (HIC) (22), affinity chromatography with mimetic ligands (19, 20), hydroxyapatite chromatography (23, 24), and other modes used in combination (25).
HCIC was introduced by Burton et al. in 1998 (26). Its mechanism is based on the pH-dependent behavior of dual- mode, ionizable, hydrophobic ligands. Details concerning chromatographic mechanism and optimization strategies are presented below and in Part 2 of this two-part report (BPI June 2015, in press). HCIC has been evaluated in schemes for purification of therapeutic antibodies that involve two or three chromatographic steps (3–5). One study describes its use in conjunction with CIEX, with evaluation of both techniques for capture chromatography (3).
In another study, HCIC was evaluated as a polishing step directly following capture on a protein A sorbent, with anion-exchange chromatography (AIEX) used as the third chromatographic step (4). The authors report that replacing conventional HIC with HCIC significantly enhanced process flow. HCIC requires no addition of lyotropic salt. Moreover, the need for diafiltration in advance of AIEX was eliminated because the product pool from HCIC was recovered in dilute buffer. Significantly, the authors report more general and straightforward HCIC optimization for antibody purification than with conventional HIC, thereby facilitating process development.
In multistep schemes, HCIC has been shown to contribute effectively to clearance of CHO host-cell proteins (CHOPs), DNA, and viruses (3). When used for capture chromatography, HCIC can provide better aggregate clearance than protein A sorbents can (4). In a related study, clearance of aggregates, CHOPs, and product- related impurities was enhanced by controlling HCIC based on both pH and the presence of binding-promoting salt in the wash and elution buffers used (5). Taken together with our findings presented below, these results suggest that HCIC could play a broader role in MAb platform development.
Materials and Methods
Chemicals, Biologicals, and Equipment: Abbott’s diagnostics division provided cell-culture supernatants (CCSs) for all experiments. They came from cell cultures performed in batch spinners, with hybridoma serum-free media supplemented by AlbuMAX lipid-rich bovine serum albumin, porcine transferrin, and insulin (all from Life Technologies). Before purification, in some cases, we concentrated the CCSs 30-fold using tangential-flow filtration (TFF) with a 50-kD Pellicon cassette from EMD Millipore to a final antibody concentration of ~1.5 mg/ mL, then 0.2 µm-filtered the resulting feed before loading it onto a chromatography column.
MEP HyperCel sorbent from Pall Life Sciences was initially packed in columns of 0.46-cm internal diameter (ID) and 5-cm height from Upchurch Scientific (IDEX Health and Science). We optimized the process for a 1.0-cm ID glass column from Omnifit Ltd. and scaled up to a 5.0-cm ID × 30-cm column from GE Healthcare. Except for the scale-up run using a PKL6 chromatography system from Pall, we ran all tests on an ÄKTA Explorer chromatography system from GE Healthcare. All chemicals used in this study came from Sigma Aldrich and were rated analytical grade.
We determined total protein content using a bicinchoninic acid (BCA) assay kit from Pierce Biotechnology. Precast polyacrylamide gels and Mini Protean II electrophoresis equipment came from Bio-Rad Laboratories. Gels were stained by SimplyBlue SafeStain reagent from Life Technologies. We performed size-exclusion chromatographic (SEC) analysis using a TSKgel G4000SWXL column from Tosoh Bioscience on a high- performance liquid chromatography (HPLC) system from Waters equipped with an autosampler and a photodiode array detector.
Column Packing: The HCIC sorbent came in slurry form with 1 M NaCl-containing 20% (v/v) ethanol ~70–75% (v/v) concentration. After a desired volume of slurry was transferred to a graduated container, we performed two decantations in 10 column volumes (CV) deionized water followed by two decantations in 5 CV packing buffer, phosphate-buffered saline (PBS) at pH 7.4 as follows:
- Sorbent was mixed with buffer or water and left to settle, then the supernatant was removed.
- A 50–75% slurry was finally prepared through addition of PBS at pH 7.4.
- A few milliliters of packing buffer were introduced into the column and drained through its lower plunger, leaving 1 cm of buffer above the bottom frit. The outlet column was closed and the upper plunger primed to eliminate air bubbles.
- Slurry was continuously poured into the column and left to settle so that a layer (1–2 cm) of clear supernatant became visible at the top of the column. The upper plunger was then adjusted so that no air bubbles were trapped.
- The outlet column was opened, and the pump was operated at a packing flow rate of 1,000 cm/h until a stable packed bed was obtained.
- The pump was then stopped and the upper plunger carefully adjusted to the surface of the bed (with excess buffer left to drain through the column inlet).
- The column inlet was reconnected to the pump without introducing air bubbles, the outlet was opened, and a flow rate of 1,000 cm/h was applied for a few minutes. If necessary, the upper plunger would be finally readjusted to the bed surface.
Chromatography: Table 1 summarizes the chromatographic conditions for all experiments. Columns were equilibrated with a minimum of 5 CV of PBS at pH 7.4, continuing until influent and eluent showed equivalent pH and ionic strength. Concentrated CCSs and unconcentrated material were loaded onto columns without preliminary adjustment. After loading was complete, columns were washed with PBS at pH 7.4 until absorbance reached baseline.
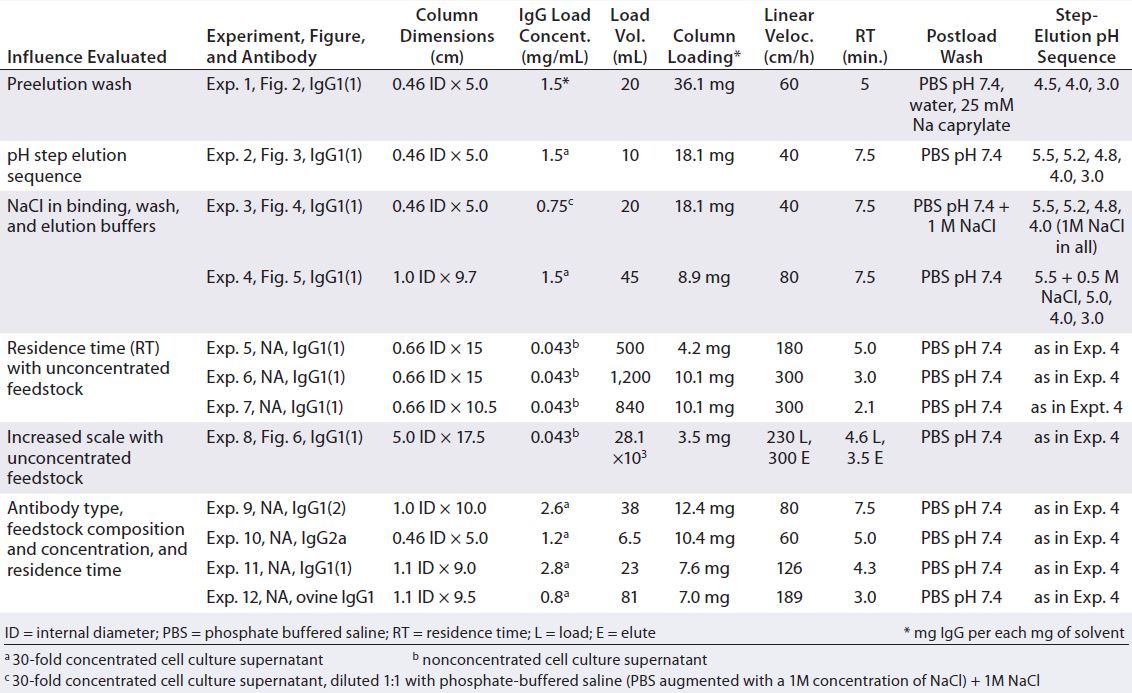
Table 1: Objectives, chromatographic conditions, monoclonal antibodies involved (all murine except for Experiment 12), and feedstock compositions; all murine experiments except 9 and 10 used the same clone of murine IgG1, designated murine IgG1(1); experiment 9 used another clone, murine IgG1(2). Experiments 1–10 used supernatant from cell culture growth medium A (a hybridoma serum-free medium (HSFM) supplemented with transferrin (10 µg/mL), insulin (10 µg/mL) and Albumax lipid-rich bovine serum albumin at a concentration proprietary to Abbott); experiment 11 came from medium B (HSFM supplemented by 0.05% F-68 pluronic acid), and experiment 12 came from medium C (Iscove’s modified Dulbecco’s medium supplemented by 10% fetal bovine serum). Figures 2–6 appear in Part 2 of this report.
For Experiment 1 only, we performed two additional preelution wash steps to prompt selective desorption of bound albumin. We used deionized water for the first wash and 25 mM sodium caprylate in PBS at pH 7.4 for the second (8). Elution involved a sequence of 100 mM or 50 mM sodium acetate buffers at different pH levels. During all the chromatography runs, we measured absorbance of effluents at 280 nm and collected fractions. For sorbent regeneration, we used a minimum of 5 CV of 1 N NaOH, and for reequilibration we used PBS at pH 7.4 or (after a longer storage period) a 1 M NaCl, 20% ethanol (v/v) solution.
Assays: To evaluate total protein content in chromatography fractions, we used a BCA assay kit following supplier instructions (27). We ran sodium-dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analyses using 15-well precast 12% (w/v) gels (28). Samples were diluted threefold in Laemmli sample buffer containing 5% (v/v) β-mercaptoethanol. We loaded 12 µL of diluted sample into each well of the gel, then applied 50 V for 10 minutes and 200 V for 42 additional minutes. SimplyBlue SafeStain gel staining was a microwave procedure, whereas destaining used deionized water. For SEC analyses, we injected 30 µL of sample diluted twofold in mobile phase (0.05 M sodium phosphate and 0.1 M Na2SO4 at pH 6.8) onto a TSKgel G4000SWXL column, then ran it at 0.5 mL/min for 45 minutes.
Results and Discussion
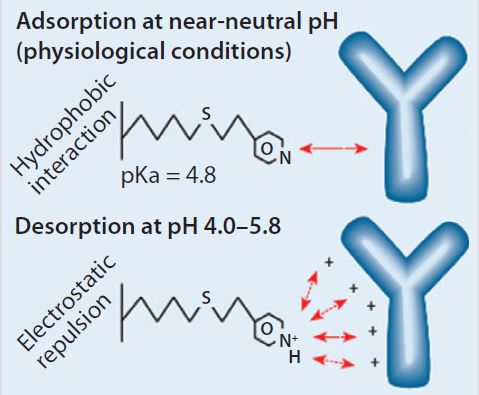
Figure 1: HCIC mechanism; binding of IgG is supported by a combination of hydrophobic interaction and molecular recognition. Desorption is prompted by electrostatic charge repulsion through reduction of the mobile-phase pH.
Our findings are best understood in connection with the mechanism of HCIC on MEP HyperCel sorbent (Figure 1). At near-neutral pH, the MEP ligand is uncharged and supports binding by hydrophobic interaction. In addition to its hydrophobic properties, the ligand is immunoglobulin selective (8, 13). A combination of affinity and hydrophobic interactions typically allows immunoglobulin binding at near-neutral pH and physiological ionic strength. Generally, no addition of lyotropic or other binding-promoting salt is required (8), although adding salt could be evaluated as a means of providing increased binding, with binding capacities <20 mg/mL observed at physiological ionic strength.
As Figure 1 illustrates, desorption is prompted by electrostatic charge repulsion, which is induced when mobile-phase pH is reduced (establishing a net-positive charge on both ligand and bound protein). HCIC optimization typically begins with determining the optimum pH for selective elution of a target antibody. To eliminate potential ambiguities associated with pH control or interpretation of findings from pH-gradient experiments, a pH-step– elution sequence is often used to determine that value. But some investigators have successfully used pH gradients during their optimization studies (3).
Control of HCIC chromatography also can be established through addition of a binding-promoting salt to elution or preelution wash buffers. Here, HCIC is controlled similarly to that conventional HIC. For example, if an impurity is found to coelute with the target antibody even at an optimum elution pH, then further optimization can be achieved by adding a binding-promoting salt to the elution or preelution wash buffer. That will prompt selective retention of either the impurity or the target antibody. We combined both control mechanisms, as described in Part 2 next month.
Finally, we investigated the influence of residence time and feedstock concentration. In addition, we compared isolation results from murine IgG1 from albumin-supplemented and albumin- free growth media. We also purified antibodies representing additional isotypes and species sources. Table 1 summarizes the chromatographic conditions, antibodies, and feedstock compositions for all experiments. It includes a summary statement of objectives for each experiment or group of experiments. In Part 2 of this two-part report, experiments are numbered according to their Table 1 listings.
References, Part 1
1 Sharkey RM, Goldenberg DM. Targeted Therapy of Cancer: New Prospects for Antibodies and Immunoconjugates. CA Cancer J. Clin. 56(4) 2006: 226–243.
2 Wild D, Davies C, Wild D. The Immunoassay Handbook. Stockton Press: New York, NY, 1994.
3 Ferreira GM, et al. Chromatography: A Two-Column Process to Purify Antibodies Without Protein A. BioPharm Int. 20(5) 2007.
4 Chen J, Tretrault J, Ley A. Comparison of Standard and New Generation Hydrophobic Interaction Chromatography Resins in the Monoclonal Antibody Purification Process. J. Chromatogr. A 1177(2) 2008: 272–281.
5 Chen J, et al. The Distinctive Separation Attributes of Mixed-Mode Resins and Their Application in Monoclonal Antibody Downstream Purification Process. J. Chromatogr. A 1217(2) 2010: 216–224.
6 Gagnon P. Chapter 9: Protein A Affinity Chromatography. Purification Tools for Monoclonal Antibodies. Validated Biosystems: Tucson, AZ, 1996; 162.
7 Guerrier L, Flayeux I, Boschetti E. A Dual-Mode Approach to the Selective Separation of Antibodies and Their Fragments. J. Chromatogr. B Biomed. Sci. Appl. 755(1–2) 2001: 37–46.
8 Guerrier L, et al. New Method for the Selective Capture of Antibodies Under Physiological Conditions. Bioseparation 9(4) 2000: 211–221.
9 MEP HyperCel Sorbent for Hydrophobic Charge Induction Chromatography. Pall Life Sciences: Port Washington, NY, 2004.
10 Boschetti E, et al. Hydrophobic Charge Induction Chromatography. Gen. Eng. News 20(13) 2000.
11 Caven TH, et al. IL-21 Dependent IgE Production in Human and Mouse In Vitro Culture Systems Is Cell Density and Cell Division Dependent and Is Augmented By IL-10. Cell. Immunol. 238, 2005: 123–134.
12 Ford JW, et al. In Vivo Murine CD23 Destabilization Enhances CD23 Shedding and IgE Synthesis. Cell. Immunol. 243, 2006: 107– 117.
13 Schwartz W, et al. Comparison of Hydrophobic Charge Induction Chromatography with Affinity Chromatography on Protein A for Harvest and Purification of Antibodies. J. Chromatogr. A 908, 2001: 251.
14 Eppink MHM, et al. Platform Technology for Developing Purification Processes. BioPharm Int. 20, 2007: 44–50.
15 Hjelm H, Hjelm K, Sjöquist J. Protein A from Staphylococcus aureus: Its Isolation By Affinity Chromatography and Its Use As an Immunosorbent for Isolation of Immunoglobulins. FEBS Letters 15 November 1972: 73–76.
16 Kronvall G. A Surface Component in Group A, C, and G Streptococci with Non- Immune Reactivity for Immunoglobulin G. J. Immunol. 111(5) 1973: 1401–1406.
17 Grönberg A, et al. A Strategy for Developing a Monoclonal Antibody Purification Platform. BioProcess Int. 5(1) 2007: 48–56.
18 Mehta A, et al. Purifying Monoclonal Antibodies. Chem. Eng. Progr. 104, 2008: S14– S20.
19 Li R, et al. Design, Synthesis, and Application of a Protein A Mimetic. Nat. Biotechnol. 16(2) 1998: 190–195.
20 Palombo G, Verdoliva A, Fassina G. Affinity Purification of Immunoglobulin M Using a Novel Synthetic Ligand. J. Chromatogr. B Biomed. Sci. Appl. 715, 1998: 137–145.
21 Necina R, Amatschek K, Jungbauer A. Capture of Human Monoclonal Antibodies from Cell Culture Supernatant By Ion Exchange Media Exhibiting High Charge Density. Biotechnol. Bioeng. 60(6) 1998: 689– 698.
22 Manzke O, et al. Single-Step Purification of Bispecific Monoclonal Antibodies for Immunotherapeutic Use By Hydrophobic Interaction Chromatography. J. Immunol. Meth. 208(1) 1997: 65–73.
23 Stanker LH, Vanderlaan M, Juarez- Salinas H. One-Step Purification of Mouse Monoclonal Antibodies from Ascites Fluid By Hydroxylapatite Chromatography. J. Immunol. Meth. 76(1) 1985: 157–69.
24 Bukovsky J, Kennett RH. Simple and Rapid Purification of Monoclonal Antibodies from Cell Culture Supernatants and Ascites Fluids By Hydroxylapatite Chromatography on Analytical and Preparative Scales. Hybridoma 6(2) 1987: 219–228.
25 Follman DK, Fahrner RL. Factorial Screening of Antibody Purification Processes Using Three Chromatography Steps Without Protein A. J. Chromatogr. A 1024(1–2) 2004: 79–85.
26 Burton SC, Harding DRK. Hydrophobic Charge Induction Chromatography: Salt Independent Protein Adsorption and Facile Elution with Aqueous Buffers. J. Chromatogr. A 814(1–2) 1998: 71–81.
27 Smith PK, et al. Measurement of Protein Using Bicinchoninic Acid. Anal. Biochem. 150(1) 1985: 76–85.
28 Laemmli UK. Cleavage of Structural Proteins During the Assembly of the Head of Bacteriophage T4. Nature 227(5259) 1970: 680– 685.
At the time this work was done, Stéphanie Chaudoreille was an R&D scientist, and Patrick Santambien was a senior R&D manager at Pall Life Sciences, 48, Avenue des Genottes, 95800 Cergy St. Christophe, France. Natalie Bohrer is a scientist in global protein sciences at Abbvie, 1 North Waukegan Road, North Chicago, IL, 60064. René Gantier is R&D director of biopharm applications, and Warren Schwartz is senior technical director of chromatography at Pall Life Sciences, 20 Walkup Drive, Westborough, MA 01581. Corresponding author Steven P. Allen is manager of biologics process design Abbott Diagnostics Division, 100 Abbott Park Road, Abbott Park, IL 60064; 1-224-668-1006; steven.allen@abbott.com.