Cell- and tissue-based therapies are being used increasingly to treat many diseases for which currently no other adequate treatment options are available. These products contain human or animal cells that can replace, regenerate, or augment a recipient’s diseased, dysfunctional, or injured cells, tissues, or organs. Cells or tissues might be unmanipulated, or their biological characteristics can be altered ex vivo before administration of the final product to patients. Examples of cell therapies range from traditional blood transfusions to recent approaches in autologous stem cell transplants and allogeneic engineered tissue substitutes.
Components used to make a cell-or tissue-based product can vary greatly in their sources, complexity, and manufacturing processes. Because such materials can profoundly affect the expansion, differentiation, and activity of a processed cell or tissue product, they can in turn have a significant impact on final product quality attributes. In addition, the biological complexity of a cell- or tissue-based therapy precludes a complete molecular characterization of the final product. Ensuring the quality of a cell- or tissue-based therapeutic thus requires rigorous evaluation of the components used in its manufacture.
Examples of Cell- and Tissue-Based Therapies: In March 2012, the US Food and Drug Administration (FDA) approved a biological license application (BLA) for a tissue-engineered allogeneic (donor-derived) cell product called Gintuit from Organogesis. Made of bovine collagen with cultured human keratinocytes and fibroblasts derived from neonatal foreskin, the product is a cellular sheet used for surgical treatment of oral mucogingival conditions in adults.
Several mesenchymal stem cell (MSC) or stromal products received approval in countries outside the United States in 2012. Regulatory authorities in Canada and New Zealand granted marketing authorization to a product called Prochymal (Osiris Therapeutics) consisting of allogeneic bone-marrow– derived MSCs for treating steroid-resistant graft-versus-host disease in children. Two MSC products are approved in South Korea: an allogeneic umbilical cord blood– derived MSC-based product (Cartistem) for treating degenerative arthritis, and an autologous adipose tissue-derived MSC product (Cupistem) for treating anal fistulas in patients with Crohn’s disease.
Component MaterialsManufacturing raw materials are defined as the starting materials, reagents, and solvents used in manufacturing therapeutic products. The quality of raw materials and intermediate manufacturing components is critically evaluated by regulatory authorities. Qualifying the supply of complex manufacturing components needed for cell therapy products can be difficult. Some components may be initially developed for research use only, so their use in manufacturing would require additional quality testing beyond what suppliers can provide as part of their certificates of analysis (CoAs). Use of components that comply with pharmaceutical quality system requirements provides assurance to cell product manufacturers and allows regulatory agencies to focus on quality assessment of therapeutic products, themselves.
Ancillary materials (AMs) — also known as ancillary products, ancillary reagents, and process reagents — are raw materials that are not intended to be present in a final product but are critically important in its manufacturing. AMs for cell- and tissue-based therapies include serum derivatives, antibiotics, cytokines, growth factors, culture media, antibodies, and enzymes. Many AMs are used to promote the growth and survival of certain cell populations and thus can have profound and long-lasting effects on a cell product. Because many AMs are themselves complex biological materials, their quality attributes may be difficult to assess. Concerns about raw-material qualifications have been amplified recently by worldwide supply chain issues such as cases of intentionally adulterated cough syrup, heparin, and infant formula (1). So it is necessary to carefully scrutinize all materials used to make cell therapies and thus prevent introduction of adventitious agents and toxic impurities while ensuring the ultimate safety, efficacy, and consistency of a final cell product.
The US Pharmacopeial Convention (USP) is a nonprofit scientific organization that develops standard tests for the identity, strength, quality, and purity of medicines and their ingredients. USP standards come in the form of monographs, general chapters, and general notices published in its compendia: the United States Pharmacopeia and the National Formulary (USP–NF). The organization also provides chemical and biological reference materials known as USP reference standards (RSs). Once those become components of applicable monographs or other compendial standards, they are used by manufacturers to conclusively demonstrate compliance with such standards.
All USP standards — whether documentary or reference material called out for use as a monograph component — must be deemed suitable by an appropriate USP expert committee. With increasing use of cell and tissue-based therapies, USP continues to develop more compendial standards for qualification of their AMs. Ultimately, assurances about AM quality are critical for cell-based therapies to meet compliance requirements. Adulteration and misbranding provisions of the US Federal Food, Drug, and Cosmetic Act (FDCA) incorporate by reference many USP–NF standards, particularly those pertaining to identity, strength (potency), quality, and purity. But USP is not responsible for enforcing any of its standards. Such enforcement is the responsibility of the FDA and other government authorities in the United States and elsewhere.
Regulations and GuidancesNo AM regulations currently exist among the world’s regulatory frameworks. General guidance about components and raw materials (including AMs) is available from several sources, however: the FDA, the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), and the International Organization for Standardization (ISO). However, their guidance documents provide limited detail on how manufacturers can develop and execute AM qualification programs. As described in the current good manufacturing practice (CGMP) regulations for finished pharmaceutical outlined in the US Code of Federal Regulations (CFR) Parts 210 and 211, AMs also can be analogous to components and product-contacting containers (2, 3). AMs are also considered “supplies and reagents” as defined by 21 CFR Part 1271 (4). So the FDA regulates cell and tissue-based products as either 361 (“minimally manipulated”) or 351 (“more than minimally manipulated”) products.
With minimally manipulated 361 products, submission of a new drug application (NDA) or BLA is not required for market entry. However, such products must comply with current good tissue practices (CGTPs), which ensure proper screening, identification, and testing of tissues and
donors to prevent introduction and transmission of communicable diseases. AMs used for 361 products do not need to be prospectively evaluated for quality. However, facilities used to manufacture such products are inspected for CGTP compliance, and records and specifications related to AMs are evaluated during inspections.
Marketing of human cells, tissues, and cellular or tissue-based 351 products (HCT/Ps) that are “more than minimally manipulated” requires adherence to more complex regulations. These products require compliance with both CGTPs and CGMPs; sponsors must file both an IND and a BLA. In addition, prospective evaluation of AM quality takes place during IND review, pre- BLA inspection, and GMP inspection. Evaluation of AM quality for 351 products is more detailed because of specific FDA requirements for AMs of human and animal origin. Robust qualification programs, identity testing, and demonstration of AM removal are just some of the critical elements used to evaluate these products.
In addition to guidelines that apply specifically to HCT/Ps, a number of general regulatory guidelines for finished pharmaceutical products also apply to cell and tissue products. Along with ICH Q7 and Q10 quality guidelines, a 2006 FDA guidance describes quality system models for examining any materials that come into contact with a therapeutic product — including AMs (5, 6). The goal of such evaluations is to ensure that suppliers provide materials that meet specifications and that manufacturers of pharmaceutical products have processes in place for control of outsourced activities and the quality of purchased materials. That includes selection of suppliers and monitoring of incoming materials. Most regulatory bodies agree on the need for building strong pharmaceutical systems to develop, evaluate, document, and monitor processes and activities. However, few regulations specify tools for enabling AM qualification.
Manufacturer ConsiderationsUnlike most small-molecule pharmaceuticals and many biologic drugs, cell- and tissue-based therapies usually are produced in uniquemanufacturing environments that include equipment and processes for culturing, testing, and packaging live cells. The initial step in developing a cell therapy requires harvesting materials from human donors — typically in a clinical setting. Harvested materials are then manipulated and/or processed in a GMP setting before being administered to patients, also typically in a clinical setting.
Characterization of cell-based therapies typically differs from that of traditional pharmaceutical products. Product purification methods for traditional small-molecule drugs are highly sophisticated and often validated to demonstrate that nothing is present in a final product other than the desired active pharmaceutical ingredient (API) and excipients. The biological complexity of cell- and tissue-based therapies precludes such definitive molecular characterization. The unique processes by which such therapies are made and tested underscores the need for careful qualification of manufacturing all components. Other critical considerations include the manufacturing environment, operator variability, and manufacturing equipment. Ultimately, a quality systems approach can be applied to every cell product component, process variation, raw-materials traceability, environmental testing for cleanroom environments, and much more.
Another major difference between cell therapy products and traditional pharmaceuticals is the number and complexity of raw materials used in final-product manufacturing. Development of cell-based therapies often requires a larger number of complex raw materials than drug products made through chemical synthesis. So it is important for a manufacturer to determine which components are most critical to quality. When added during cell product manufacturing, some raw materials (e.g., AMs such as cytokines and growth factors) can affect cell growth, differentiation, or function. Any variability in AM quality can affect the quality of a final cell therapy product. Thus, in assessments of qualification needs, AMs are considered more critical components than a raw materials that do not play a role in cell growth, differentiation, or function.
As Figure 1 shows, different measures can be taken to help ensure quality as cells progress through different stages of product development and manufacturing. When a manufacturer submits a cell therapy product dossier for regulatory approval, a regulatory agency may request information about qualification of AMs used in making the final product. If the manufacturer has not qualified AMs throughout its development process, then it may need to request information from the AM supplier about its own qualification program. If such information is limited or unavailable, then the regulatory approval process could be hindered significantly.
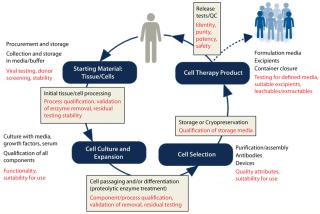
The nature of an AM’s manufacturing process is a keyconsideration for cell product manufacturers. Some AMs actually may be approved pharmaceutical products in themselves. Heparin and insulin, for example, are available as GMP-grade materials and often used in cell product manufacturing. But end users still must determine whether pharmaceutical-grade AMs demonstrate quality attributes appropriate to their use in cell product manufacturing. In addition, drugs often use stabilizers or other additives that are not desired in cell culture.
Risk-Based Approach to Ancillary Materials (AMs)
Tier 1: Low-risk, highly qualified materials with intended use as therapeutic drugs or biologics, medical devices, or implantable materials
Tier 2: Low-risk, well-characterized materials with intended use as AMs, produced in compliance with GMPs
Tier 3: Moderate-risk materials not intended for use as AMs (frequently produced for in vitro diagnostic use or reagent-grade materials)
Tier 4: High-risk materials, those not produced in compliance with CGMPs and not intended for use in cell manufacturing
When assessing risks associated with using AMs during cell therapy development, manufacturers should weigh the benefits of using qualified material (e.g., USP-grade material) early in product development. Introducing well-qualified materials earlier in development will lower risk by eliminating the need to change suppliers later in a product’s life cycle. Because cell- and tissue-based products can persist or propagate in patients long after administration, it is a reasonable goal for all product-contact materials to be properly tested or qualified at all stages of product development.
USP’s Standards As ToolsAn Overview of Ancillary Materials: As described above, qualifying cell-based therapies involves unique challenges. USP general chapter <1046> provides information on good practices to overcome those challenges in manufacturing, characterization, and testing of HCT/Ps (7). The chapter also discusses potential impacts of AMs on final product quality.
USP’s informational general chapter <1043> is another tool available to manufacturers interested in developing appropriate AM qualification programs (8). This chap
ter discusses what qualification programs for ancillary materials should address: lot-to-lot and vendor-to-vendor variability, traceability, suitability of use, impact on quality and safety of finished products, and other critical parameters. It describes critical components of identification, which includes source material, concentration of use, tests for identification, and manufacturing steps. The chapter describes the importance of AM selection in an early phase of development as well as microbiological assessment and risk assessment with animal-based products (e.g., those associated with transmissible spongiform encephalopathies). Chapter <1043> is currently being revised to incorporate more information of potential use to manufacturers.
As the “Risk-Based Approach” box shows, general chapter <1043> emphasizes the importance of proper AM selection and describes four risk-based categories for classifying critical materials. Tier 1 includes low-risk, highly qualified material such as approved pharmaceutical products (e.g., pharmaceutical-grade heparin). Tier 2 AMs are low-risk, well-characterized materials intended for use as AMs. Tier 3 includes materials with moderate risk, often not intended for use as AMs (e.g., reagent-grade materials used in other applications or industries). High-risk materials are categorized as Tier 4. They are not produced in compliance with CGMPs or intended to be used in cell therapy manufacturing. Based on these categories, companies can devise qualification or risk-reduction activities for their AMs. Table 1 provides an example of Tier 1 AMs, listing their uses in manufacturing and proposing qualification or risk-reduction activities.
Table 1: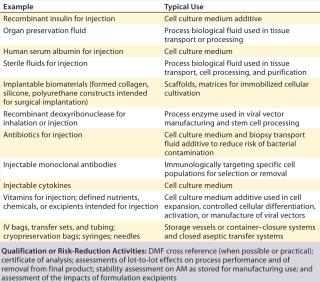
Table 2provides even greater detail for qualification levels and describes risk-reduction approaches that need to take place once a risk assessment is complete and critical components are identified. As cell therapy productAMs move from the lowest to the highest risk level, the amount of work associated with their risk assessment and risk reduction increases. In particular, when AMs are derived from human or animal sources, adventitious-agent traceability and control are critical elements in risk reduction, and mitigation of some key elements needs to be addressed.
Table 2:
USP’s Approach to AM Standards: Building on general information from chapter <1043>, other general chapters in USP–NF provide greater detail about conducting tests of quality attributes associated with specific types of AM products used in cell manufacturing applications. Such chapters include <90>, <92>, <130>, and <1024> (9,–14). Test specifications in those chapters are based on process consistency in AM manufacturing and validated test methods. In addition, USP provides physical RSs that can be used by manufacturers in testing their products and AMs according to USP documentary standards. Those RSs can be valuable for developing methods to detect residual AMs in final products.
Upcoming Workshop
On 7–8 November 2013, the USP and the International Society of Cellular Therapy (ISCT) will host a workshop in Rockville, MD: Cell and Tissue-based Regenerative Medicine Products: From Characterization to Compendial Assays. It will bring together key stakeholders from industry, academia, and government to identify opportunities and challenges in defining characterization assays for cell and tissue-based products as well as establish priorities and identify assays ready for inclusion in the United States Pharmacopeia—National Formulary. For details about the workshop and registration, go online to http://uspgo.to/cell-tissue.
Use of animal-derived AMs is discouraged in manufacture of therapeutics for human use, not only because of a risk of animal–human disease transmission, but also because of the potential for immunogenic reactions to animal proteins. The latter is a major concern when products are used in repeat dose regimens. In the absence of a suitable alternative to animal-derived AMs such as fetal bovine serum (FBS), cell product developers must use materials that have been evaluated using strong qualification programs to meet acceptable test specifications. USP general chapters <1024> and <1043> provide valuable tools for addressing traceability approaches and qualification strategies, respectively, whereas chapter <90> provides tools for assessing critical quality attributes for a material such as FBS. The criticality of AM quality attributes can depend on the application for which an AM is used. Testing an AM against a USP RS ensures confirmation of its identity and functionality.
The USP is developing standard testing procedures and reference materials for a number of recombinant growth factors and cytokines that are commonly used in cell culture manufacturing: e.g., interleukin-4 (IL-4), granulocyte–macrophage colony-stimulating factor (GM-CSF), tumor necrosis factor alpha (TNF-a), and FGF2 and EGF growth factors. USP general chapter <92> addresses some cytokines and growth factors (IL-4 and FGF2) and will include others in future revisions. The USP RS for IL-4 was developed in a multilaboratory collaborative study intended to assign the RS material a specific activity using a cell-based assay. Units were calibrated against an existing World Health Organization (WHO) international standard for IL-4. Using material calibrated against USP’s standard can help a company ensure process consistency and use the right amount of material for each cell culture application. In addition, by using the USP-described test where it is specified in an applicable compendial standard, users also will be assured of a material’s identity.
Multiple Sources for InformationAs our understanding of complex diseases and disorders grows, the search for better ways to address them will continue. An expanding body of research on cell- and tissue-based therapies is paving the way for new therapeutic products. Given the range of such treatments available today, quality considerations must take into account the qualification of the raw materials that go into their making — in particular, AMs that affect cell growth but are not intended to be present in a final therapeutic product. Although regulatory guidance currently offers manufacturers an overview of best practices regarding qualification of cell- and tissue-based pro
ducts and their components, the USP also provides manufacturers with tools for designing and developing qualification programs that specifically address ancillary materials.
Author Details
Corresponding author Fouad Atouf, PhD, is director of biologics and biotechnology with the US Pharmacopeial Convention, 12601 Twinbrook Parkway, Rockville, MD 20852-1790;