The pressure to contain rising healthcare costs — combined with the number of innovator biologic drugs coming off patent (30 licensed biological drugs by 2015) — offers huge opportunities for developers of biosimilar products. In 2011, the global market size of the biosimilars industry was around US$2.5 billion. Global demand for such products — and monoclonal antibody (MAb) biosimilars, specifically — is estimated to grow at 8–17% from 2012 to 2016 (1). The advent of biosimilars should bring more affordable biologics to market: Estimates of their costs range as much as 50–75% that of innovator biologics. That is especially important news to some developing countries where biopharmaceuticals have yet to become available.
Unlike a generic small molecule, however, a biosimilar product cannot be considered an identical copy of its innovator counterpart. Even very small differences in cell lines or manufacturing processes can have large impacts on potential side effects observed during treatment. Two “similar” biologics could trigger very different immunogenic responses. Thus, substitution of a reference biologic with a biosimilar could have significant clinical consequences. That creates safety concerns among regulators. So in recent years, a number of biosimilar guidance documents have been released by regulatory agencies that begin to address those concerns.
Europe acknowledged the requirement for regulation of biosimilar therapeutics in 2005 with publication of the European Medicines Agency (EMA) Guideline on Similar Biological Medicinal Products(2). Its content borrowed heavily from the EMA’s earlier Note for Guidance on Biotechnological/Biological Products Subject to Changes in their Manufacturing Process(3). Then in early 2012 (4), the US Food and Drug Administration (FDA) released a draft guidance document on scientific and quality considerations in demonstrating biosimilarity to a reference product (the innovator’s original biologic).
Unlike small-molecule drugs, these products are extremely complex. No single analytical/preclinical test or clinical study can demonstrate that a biosimilar is sufficiently similar to a referenced innovator biologic. One limitation of those regulatory guidance documents is that they do not actually specify how similar a biosimilar must be to the innovator’s product with which it is intended to compete. Demonstration of biosimilarity is left up to companies developing such therapeutics.
EMA and FDA guidance documents suggest that a multifactorial, stepwise approach should be taken to demonstrate biosimilarity. The FDA’s approach proposed lists steps that should be considered in generating a complete picture of how a biosimilar compares with a reference innovator, including structural and functional characterization, animal studies, and (most important) clinical studies. The agency refers to overall data generated from those assays and studies as “totality of the evidence.”
The FDA guidance does not specify that all types of testing performed on an innovator product should be repeated for the related biosimilar. Instead, the agency advocates taking a risk-based approach to biosimilars, with sufficient scientific justification provided for all testing not performed. With this approach, in vitro functional characterization will become critically important at a very early stage of development because such assays predict how a biosimilar may behave in vivo.
Biosimilar MAbsTherapeutic MAbs achieve their effects either directly by inducing apoptosis or indirectly by inducing antibody-dependent, cell-mediated cytotoxicity (ADCC) or complement-dependent cytotoxicity (CDC). The mechanism of action generally involves an antibody’s Fab (antigen-binding) and Fc (crystallizable) regions. The former binds to antigens; the latter bind to Fc receptors found on monocytes, macrophages, and natural killer cells.
Evaluating a MAb’s binding affinity to different types of Fc receptors is important for predicting its therapeutic potential. Specifically, analyzing Fc gamma receptor (Fcγ) binding determines an antibody’s ability to recruit immune cells, including natural killer cells: neutrophils and macrophages. Analyzing neonatal Fc receptor (FcRn) binding predicts the antibody’s in vivo half-life.
In 2010, the EMA issued a draft guidance on similar biological medical products containing MAbs (5). This document is specific about the tests that should be conducted on MAbs to assess the in vitro pharmacodynamics of a biosimilar relative to the innovator MAb. It states that affinity-binding to all Fc receptors should be assayed. However, there are so many Fc receptors with different alleles, some of which have little or no binding activity, that testing them all would be impractical. Submissions to European and Japanese regulatory agencies that focus on high-binding Fc receptors — including CD16a, CD32a, CD64 — have allowed companies to progress with clinical studies as part of the registration process.
EMD Millipore has developed two different methods that measure the binding of any therapeutic antibody to any Fc receptor. Surface plasma resonance (SPR) measures antibody binding to recombinant soluble Fc receptors. Flow cytometry measures antibody binding to cells that express certain receptors. Both methods have been formatted as parallel-line assays and demonstrate high levels of accuracy, precision, and linearity. That makes them valuable for use in comparability, potency, and stability assays. These methods also show greater precision and reproducibility than traditional cell-based assays that test for ADCC. Additionally, both are readily able to detect structural (e.g., glycosylation) differences between two MAbs that can affect molecular function.
In Vitro Functional TestingFcγ Receptor Assays: To measure Fc binding response, SPR assays are developed by covering the surface of a laboratory chip with recombinant human Fcγ receptors. Independently prepared serum dilutions of test samples are then injected over two flow cells — a control cell and the Fc receptor–coupled cell — and the difference in binding response is then measured.
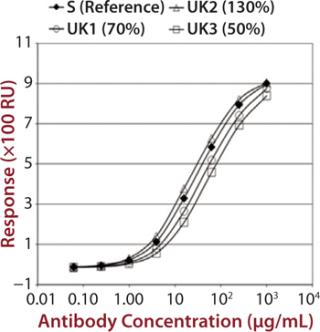
To demonstrate that such an assay could detect differences in measured binding and different potencies, we qualified it by adjusting concentration of alemtuzumab (Campath from Genzyme Corporation) from 50% through 150% of a reference concentration. Binding curves were obtained for high-binding Fc receptors CD16a, CD32a, and CD64 (Figure 1). We observed similar binding curves for all three receptors, but the magnitude of the signal obtained was markedly different for each, reflecting the MAb’s binding strength to the different receptor forms, with its highest affinity being for CD16a. After plotting the correlation between CD16a binding curve data and nominal potencies, we determined relative potency using a five-parameter logistic parallel-line model (data not shown). All curves of different starting concentrations were determined to be parallel to the reference curve (p = 0.05) for all three receptors.
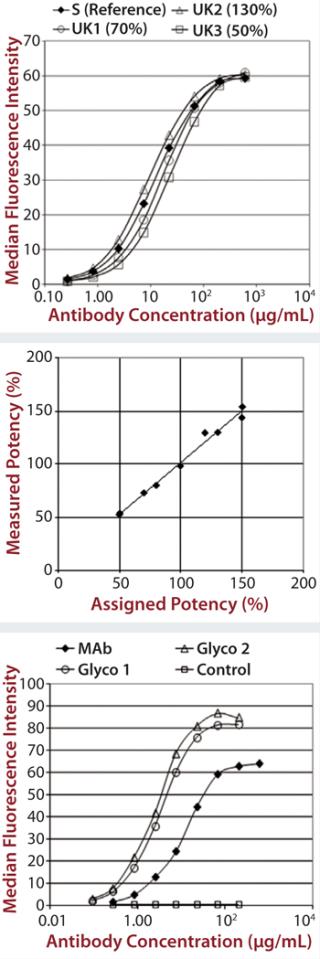
Control antibodies were then engineered either not to bind (negative control) or with enhanced binding to CD16a (Glyco1 and Glyco2), and we obtained binding curve data using SPR (data not shown). Then we qualified the assay using additional therapeutic MAbs, including bevacizumab, rituximab, and eculizumab (a negative control because it lacks Fc-receptor binding) (data not shown).
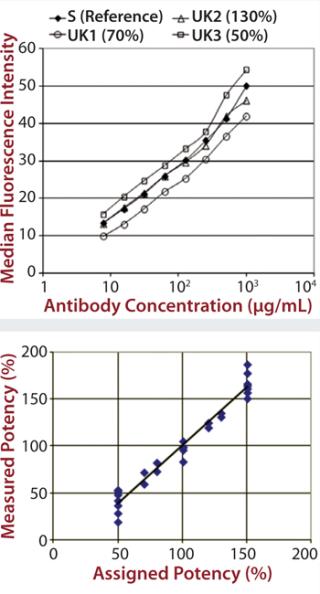
Each of those assays was reproduced by transfecting CD16a, CD32a, and CD64 into Chinese hamster ovary (CHO) cells, incubating with a therapeutic MAb and measuring the level of binding using flow cytometry. We qualified the flow cytometry assay using alemtuzumab with the same nominal concentrations to mimic different potencies of 50–150% of the reference. Assay performance and binding curves were very similar to that observed for SPR (Figure 2).
The Neonatal Fc Receptor (FcRn) Assay: The FcRn transports immunoglobulins across the placenta between mother and fetus. It is also responsible for salvaging internalized immunoglobulin and albumin, thus making it responsible for the long in vivo serum half-life of MAbs. As specified in the EMA biosimilar draft guidance (5), consequently, neonatal Fc-receptor binding must also be tested for biosimilar MAbs.
Unlike other Fc receptors, the FcRn protein is not commercially available. So EMD Millipore manufactured a supply of recombinant dimeric receptor suitable for use in SPR binding assays. Our FcRn assay format is very similar to that used for the Fcγ receptors, except that the FcRn protein is coupled directly to the chip surface rather than coupled to it by a histamine tag. Although the actual order of magnitude for the signal obtained was low compared with the Fcγ receptor results (Figure 3), the data were consistent and reproducible.
C1q Assay: The final assay we developed to satisfy the requirements of the EMA biosimilar antibody guidance measures a MAb’s binding to C1q. That receptor is the first component of the complement cascade; hence, it is very important in the mechanism of action for therapeutic MAbs. C1q is a very large, hexameric molecule, and its binding affinity in solution is extremely low, making it difficult to measure. Although it is possible to measure C1q binding to immobilized immunoglobulin G (IgG), that would be difficult to do in an assay that is suitable for potency or comparability studies. So EMD Millipore developed an alternative approach using an enzyme-linked immunosorbent assay (ELISA) method (Figure 4).
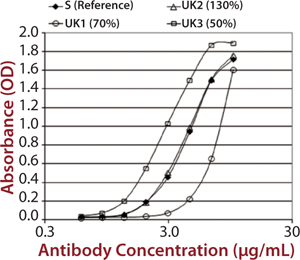
Figure 4:
Binding to soluble or cell-surface Fc receptors can be accurately measured using SPR and flow cytometry, respectively. Both techniques have excellent precision for replications (typically <5% CV), and their results are amenable to potency determination according to the parallel-line method. Additionally, they can easily distinguish between variant glycoforms of an antibody and between different antibodies, providing good correlation with the results of cell-killing experiments without their associated variability.
These assays could be sensitive to changes in antibody function related to glycosylation, aggregation, concentration, and protein modification. The techniques can be applied to measure antigen-binding activity of therapeutic antibodies as well as many other drug–ligand interactions. Combining them with target binding can provide a comprehensive picture of functional activity that is sensitive to small changes and could thus provide a reliable tool for quality control and comparison of biosimilars with innovator products.
This is just one very small part of the larger picture with biosimilars, however. Many changes can occur in biological molecules without affecting their potency or functional data. Thus, careful physicochemical characterization is still essential (as well as preclinical and clinical testing) to ensure a biosimilar’s comparability with its referenced innovator product.
EMD Millipore has recently completed biosimilar testing using the assays described herein. Most of the Fcγ, FcRn, and C1q assays gave results within 10% relative potency of the innovator biologics. These data were submitted to European regulatory agencies, and approval for a clinical trial was granted based on these and other data in the dossier.
Author Details
Corresponding author James Hulse, PhD, is managing scientific director of discovery and development solutions North America for EMD Millipore. Chris Cox, PhD, is a freelance bioanalytical laboratory consultant working for Merck Millipore.