At the IBC Third Annual International Forum on Vaccine Production, I presented an outline of “Best Practices for Quality by Design (QbD) in Biological Products and How to Implement in Vaccines.” It covered process development and QbD principles, best practices used in biologics, how QbD fits in with process validation, how it applies to vaccines, and some thoughts on the potential for seasonal vaccines.

Classic process development (as practiced in the early days) generally involved rudimentary processes that were “patched together.” By performing sequential, single-parametric analysis, you could define general processing parameters. Generally, little attention was paid to the impact of raw materials unless a disaster occurred. Because of the limiting availability of resources and time, development was sparse and not fully complete. Then by rigorously controlling process parameters, operational staff hoped and prayed that the ensuing product was acceptable. In many situations, it was not. So parameters were tightened further in hopes of solving the problems. In those early days, the end result was low efficiency, poor reproducibility, and a high failure rate in production.
Over the past 20 years, however, things have begun to change. Our experimental design strategy has evolved — allowing us to design experiments to test the influence of multiple parameters simultaneously and efficiently. We have learned to analyze data statistically and ascertain whether a parameter is significant or insignificant. We now recognize that the quality of raw materials can and does have the potential to affect the outcome of manufacturing and that specifying “USP or EP grade” is not the answer. Some raw materials do not need to be USP or EP grade, but others must meet more stringent requirements. We are beginning to scratch the surface of the impact.
Similarly, we have begun to recognize that not all parameters and points in the process have the same impact on the outcome. Some critical control points must be rigorously monitored and controlled to assure acceptable products. We are also beginning to understand what outputs in our unit operations are important. But these properties, referred to as critical quality attributes (CQAs), are not as clearly defined as you might think.
A QbD ApproachBetween 2005 and 2012, the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) issued quality documents Q8 and Q11 that provide guidance on how to develop products and processes to incorporate those new approaches (1, 2). Those documents describe the key elements of QbD. The key steps in the new paradigm for development are
-
Define your product profile — what your product should look like exactly and what it should do.
-
Define your CQAs for the product and critical in process steps; those attributes, when met, will yield a product that you desire in a reproducible manner.
-
Define what elements of your process (inputs and control points) need to be controlled to ensure that you attain your CQAs.
-
Determine what the operating ranges of such elements are to consistently yield acceptable product.
-
With those operating ranges, define your design space so that if you operate within it, good product ensues.
Previous articles I’ve written illustrate those concepts in greater detail (3, 4). Figure 1 shows the interplay of those inputs. Each process unit in the diagram is a unit operation, and they are linked such that the output on one is the input for the next all the way from the beginning to end (drug product).
CQAs are requirements that must be met for a process to yield an acceptable product. This is a key, relatively new concept in the industy. For a drug product, such parameters are made up of key specifications as well as other attributes that might not be specifications. Because vaccines are categorized as biological products, certain attributes might include immunogenicity or antigenicity. For other biologics, those are not necessarily desirable traits. Vaccines pose a special challenge because they are administered to healthy patients and often pediatric populations. So safety is a critical attribute — more than it would be for a biologic to treat cancer in a middle-aged patient, for example.
CQAs may take different form upstream in a process. Yield and processability downstream play important roles, as do product stability and absence of interfering or contaminating components. In spite of the different focus, the end result must be to ensure that the ultimate CQAs are met: those of the drug product.
Implementing QbDDevelop your product profile (5,6,7,8) and define CQAs unit operation to unit operation starting at the drug product and working backward. Develop your process train by understanding what is done and (just as important) what is not accomplished at each step. With that knowledge, you can fully understand the impact of a unit operation on the one before and the one after.
Then you can determine which raw materials are critical versus those that have low impact. Doing so requires sophisticated experimental design and the heavy use of quality risk management as defined in ICH Q9 (9). All of that statistical evaluation is generally done at a small scale. However, it works only if you have confidence that the small-scale model is predictive of the large-scale and you test within attainable ranges for the large scale. Thus, success here requires a validated scaled-down model for each unit operation. From this analysis, you will determine the relative importance of different raw materials and what properties you must control.
As with raw materials (or inputs), you must determine the locations of your critical control points. Those points have the potential to alter the outcome of your operations, so they must be monitored and controlled. Again, the use of sophisticated experimental design and ICH Q9 are critical.
I am not going to go into detail on the ICH Q9 document, but it is critical to understand to effectively and efficiently perform a modern QbD program. Basically, the concepts in ICH Q9 are designed to he
lp companies investigate — based on prior knowledge, experience, and experimentation — which parameters are critical (thus needing to be controlled) and which are benign and need limited oversight. Techniques such as hazard and critical control point (HACCP) analysis and failure mode and effect analysis (FMEA) are critical and useful. They can help you identify key elements so that you can focus your resources on those more and less on their less-critical counterparts.
Defining Design Space: After those studies, you should end up with a list of what does or does not need to be monitored and controlled. For parameters needing control, you also end up with the interplay between parameters and how they affect the outcome. An output of this is the design space (Figure 2), a collection of areas within which you can reliably produce products that meet your CQAs. Although you can operate within the whole design space, you may choose to operate within a smaller range (batch process setting) to further control the economics of your process. If you deviate from the settings but within a design space, a product is acceptable and as such can be released. Because your design space is included in your filing, you do not need to seek regulatory approval for those changes.
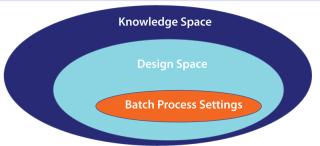
Outside a design space, there is a knowledge space. Wander outside this boundary, and you need to submit additional information to the US Food and Drug Administration (FDA). A product may be acceptable or may include edge-of-failure elements. It is very important to understand what is beyond the horizon. If you have an edge-of-failure element close to a design space, it would warrant you to keep well away.
Most of the success for QbD has been in manufacturing low–molecular-weight pharmaceuticals. Out of 35 submissions from companies processing such drugs, about six have had successful submissions. On the other hand, only six biologic submissions have been made so far (none of which are vaccines), and none have been approved to my knowledge. The successful few have been exempted from preapproval inspections and have simplified operations for internal change control instead of having to make complex submissions for process changes. Two further advantages that are in the process of being realized are improved operations and more efficient technology transfers.
Challenges to QbD implementation include addressing the following:
-
Changing the way we approach process development by incorporating new approaches of ICH Q9 and experimental design
-
Thinking for the future — establishing customer–client relationships and defining product properties and CQAs
-
Initiating and adhering to product timelines; don’t start too early or too late
-
Realizing that process development is never over; it does not stop after technology transfer, but continues throughout a product’s life cycle.
In particular, technology transfer is not the “toss it over the wall” approach of the past. Rather, it must be the process of shepherding a product into manufacturing, ensuring that a process works and that it operates smoothly with reproducibility and predictability.
ICH’s Q10 quality management system document also plays a critical role (10). This document is intended to complement good manufacturing practices (GMPs) by introducing elements from the International Organization for Standardization (ISO). In that system, management is actively involved. You have a robust change-control process linked to a product’s history file, thus embracing continuous improvement and robust quality vision and processes.
QbD and Process ValidationElements of QbD are integrated in the new paradigm for process validation (PV). The FDA and European Medicines Agency (EMA) both have issued new guidances covering PV in the past two years. Those documents have been discussed in several articles (11, 12). Gone are the old ways of transferring the process: running three batches, writing a report, and putting it on the shelf to gather dust. The new paradigm is to recognize that PV is not a one-off activity but, rather, is integrated into the development and operational life cycle of a product. Three key elements are included in a modern PV strategy:
-
Build your process; use new or QbD principles at small scale.
-
Confirm your process during scale-up; after technology transfer, confirm it is operating at full scale.
-
Continuously monitor your process in real time; evaluate how well it is running after the initial phase, and make adjustments and improvements based on your qualification program.
Perhaps the old three-run model is no longer relevant. Most biologics manufacturers recognize that three runs really cannot tell you whether a process is stable or predictable. Should we do perhaps 20? If so, do we do 20 before submission? No! FDA advocates submitting a plan to perform a relevant number after licensure, and it is valid to submit a report to confirm or verify your process development at small scale.
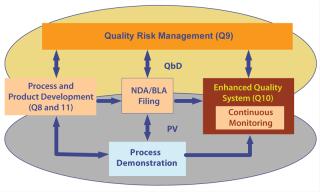
Initial verification and eventually continuous monitoring would involve examination of all critical control points — including raw materials — to ensure that a process is running predictably and consistently. During such analyses, we can question the need for change within a design space and implement it through an internal change control. Likewise, we can introduce new technology if we can maintain parameters within a design space again with internal change control. Figure 3 illustrates how we might tie all such elements together.
Integrating PV and QbD into Vaccines Development and Operations: Vaccines are a mixed bag of different products, from very modern types such as recombinant protein subunits cloned into vectors (e.g., Hep B) to complex mixtures of agents produced by old technologies. Recombinant vaccines are very much like recombinant therapeutic proteins and as such fit the standard model for PV and QbD.
Other vaccines are processed similarly and use cloned (or at least purified) components. Those also fit well. Vaccines such as inactivated whole organisms and subunits of organisms, however, are not purified components in the same way as recombinants. Such products pose special challenges with respect to defining the elements of QbD and will make the execution of QbD strategy difficult. Bearing in mind the slow implementation of
recombinant protein development into QbD, I believe that developers of such vaccines will take a “watch and wait” strategy. Their first foray into QbD might be for established products, with which there might be an advantage to submit a partial QbD plan to aid in process change for troublesome unit operations (e.g., aseptic operations or others that introduce new technologies).
Seasonal (trivalent) vaccines manufacturers have an interesting opportunity as well. Each year flu vaccine manufacturers wrestle with new strains (three strains each new season). Those strains are often difficult to grow, and a limited time to market can create a scramble. Furthermore, manufacturers have to assess and ensure that the components are antigenic and safe. Using a QbD strategy might help “validate” a production platform and identify key critical process parameters and the roles of raw materials that govern yield, antigenicity, and other critical properties. With that knowledge, manufacturers might be able to streamline their introduction of new products each year.
Such strategy is akin to what monoclonal antibody (MAb)development companies do now. They establish a standardized platform for introducing a new molecule into the clinic. Thus, the introduction of a new MAb into clinical manufacturing relies on specialized knowledge of the properties of a new molecule (e.g., stability, isoelectric point, tendency to aggregate). With that knowledge, manufacturers can make adjustments to their standard platforms for cell culture and purification. That means reasonable yield of a satisfactory molecule quickly — first in human as rapidly as possible.
With the movement from eggs as the growth environment to cell culture for flu vaccines, such a process might also aid in the implementation of a platform concept. That is, if regulators look favorably on the concept.
Author Details
Peter Calcott is president of Calcott Consulting LLC; 931 Mendocino Avenue, Berkeley, CA 94707; 1-510-316-5700;