Compliant companies, to paraphrase Tolstoy, are all alike. Every noncompliant company seems to find its own way to fall short of compliance with good manufacturing practice (GMP) and come, as did the writer’s famous heroine Anna Karenina, to grief. One commonality of compliant firms is that most seem to have excellent self-auditing/self-inspection programs. Indeed, many inspectors say that a primary predictor of a compliant company is a rigorous self-inspection program. Such a program is appropriately focused, adequately resourced, taken seriously by management, and not only discovers incipient compliance problems, but follows up by implementing effective corrective and preventive actions (CAPAs). Another common feature of happily compliant companies is an excellent regulatory intelligence (RI) system that provides the knowledge needed to stay on track in the dynamic regulatory environment
In a previous article (1), I provided a primer on lessons that can be learned by studying and analyzing FDA notices of deficiency. These are commonly known as 483s because during an inspection, the inspection team members note their observations on Form FDA-483. Here, my goal is to provide a guide on how the “appropriate focus” of self-inspections can be achieved and enhanced by setting up an RI subsystem to facilitate recognizing, tracking, and ultimately predicting inspection trends. This is presented as a three-step process: information gathering, information mining, and transformation of knowledge into action.

Gathering the Needed Information
“When action grows unprofitable,” wrote the novelist Ursula Le Guin, “gather information.” The first step toward setting up your inspection trend spotting, tracking, and predicting system is to obtain the necessary information, data, and intelligence. This process differs for each regulatory authority, and time and space constraints preclude covering each of those separately.
United States: Inspectional information from the US FDA is relatively easy to obtain as a result of the Freedom of Information Act (FOIA) as well as transparency initiatives undertaken by the agency. Productive sources include warning letters (WLs), establishment inspection reports (EIRs), and forms 483. Each of those sources has inherent advantages and disadvantages, but all are well suited for providing the RI needed to feed the inspectional radar system.
Warning letters are the most readily available source of FDA inspectional information. Every WL is placed on the agency’s website as soon as it becomes releasable under FOIA (see Table 1). WL Web pages can be searched according to date, subject, company, issuing office, and those with response and closeout letters. WLs tend to focus on violations that are the most serious and most important to the FDA. Although WLs are a rich source of inspectional RI, they have some inherent disadvantages, including a lack of timeliness and a limited scope. They are not released until they are closed out, which may not occur until years after an inspection. And because they are issued for a small percentage of inspections, WLs provide only a limited view of recent noncompliance observations.
Table 1: Web-based sources of inspectional information
Because of those limitations and because WLs have advantages of their own, FDA notices of deficiency (483s) are also an essential component of an inspectional RI system. There is still a lag time between an inspection and release of the associated 483, but it is generally much shorter than that for WLs. For example, the FDA inspected Intellicell Biosciences between 8 November and 12 December 2011, and the 483s became publicly available on 4April 2012, a lag time of less than four months. The most important and most requested 483s are posted on the FDA Office of Regulatory Affairs (ORA) website and are as readily available as WLs (see Table 1). Form 483s also cover as wide a range of inspection results as is publicly available, which makes them crucial for focusing compliance efforts.
One disadvantage of using 483s as a resource is that too few are posted on the FDA’s website. So they must be obtained through other means. The least expensive but most time-consuming method is to make a request to the FDA’s FOI office. For this, you must know (at a minimum) the names and locations of companies whose inspections you want to track, the date range, and FDA project areas. Those can be found in the FDA’s inspection database (Table 1). The front page of the database contains instructions for its use.
Once you have the necessary information, the next step is to make a FOIA request to FDA following the agency’s instructions on that subject. Some costs will be incurred because FDA charges for search and reproduction, but such costs usually amount to <$100 per search. If you conduct many searches per year, those costs can add up to more than using a newsletter subscription.
The simplest and fastest way to get 483s that do not appear on the FDA website (which is the vast majority) is to subscribe to a newsletter that publishes 483s regularly. Such newsletters charge yearly subscription fees, and although getting inspectional information in this manner is quick and easy, you do give up control over which companies and which inspections you track. Examples of newsletters that include 483s in their coverage include BioQuality, GMP Trends, and Inspection Monitor.
A final method for obtaining information about Form 483s is to use a commercial source. If information about 483s is in a company database, you can have an electronic version within minutes after paying for the documents you want. Expect costs to range from $50 to $200 or more, depending on the number of pages in the document you are purchasing. Examples of such companies include FOI Services and FDA News. The former also will conduct custom FOI requests for a fee that can run into the hundreds of dollars. As for any FOIA request, the waiting time for the document you want can be several months.
For even more inspectional detail, Establishment Inspection Reports (EIRs) can be obtained through FOIA or commercial sources such as those named above. In addition, some EIRs make their way to the FDA website. To check for that information, visit the ORA section.
Europe and Beyond: Inspectional information from Europe is much more difficult to obtain than from the United States. The European Medicines Agency (EMA
) keeps results for inspections of centrally authorized products, but — except in rarely released summaries — such information is generally not publicly available. For example, the most recent summary information was released five years ago and covers up to 2006. Some additional information can be obtained from EMA’s Inspection Page, but even the combined information from those two sources is too meager for effective inspection tracking.
In addition to EMA, each European National Regulatory Agency keeps information on inspectional results. But keeping up with the more than two dozen of such results is tedious and will daunt all but the most dedicated RI detectives. The British Medicines and Healthcare products Regulatory Agency (MHRA), however, does an admirable job of providing yearly summaries.
Even with MHRA data added in, there will probably be too little information for effective European inspectional trends tracking. Therefore, the diligent will want to supplement their knowledge with attendance at industry meetings where European inspection results are discussed. Amongthe many available meetings, those held by the British Pharmaceutical and Healthcare Sciences Society (PHSS) have been valuable, as have those of the California Separation Science Society (CASSS), especially WCBP and some of their European Forums. In addition, the professional association PDA has an inspection trends interest group (Table 1)
20 Questions: Self-Inspection Checklist for Additions to a Media Fill*
This is not meant to be a comprehensive checklist for auditing process simulation media fills but a brief example of how inspectional information can be used to augment an existing audit list prepared from regulations, guidance documents, and an auditor’s own experience.
- Does the media-fill SOP or other official written document contain instructions for establishing reconciliation criteria?
- Have criteria been established for reconciliation of filled units, including total units evaluated/incubated compared with the total number of units filled?
- Are discrepancies between the amount of units filled and the number of units evaluated/incubated justified in writing?
- Are believable rationales provided for all discarded units?
- Do quality assurance (QA) personnel review and approve the justifications and rationales?
- Does the media-fill SOP or other official written document require that all deviations be justified and that the justification be approved by QA?
- Are all interventions that occur during aseptic-fill processes incorporated into the media-fill process simulations?
- Do all operators, and not just some, perform SOP-mandated interventions?
- Are media filled vials in the area of the interventions incubated?
- If multiple interventions occur simultaneously during routine aseptic filling, are multiple simultaneous interventions included in the media fill process simulations?
- Is it a requirement that requalification including successful media fills must be performed before changes such as modifications to the vial filler and restricted access barrier are approved?
- Are investigations required, performed, documented, and reviewed for filled vials rejected during process simulations for such things as fibers and particulate matter in them?
- Is a time frame specified for these investigations?
- Is second operator verification required and performed for the number of vials filled, the number of vials to be incubated, and the number of vials actually incubated?
- Have aseptic media fill simulation lots been invalidated without justification?
- If appropriate, are product impact assessment following media fill failures adequate to assure sterility of products filled since last successful simulation by documenting that such things as sterility, release testing, complaints, and adverse events of all affected lots were reviewed to assure sterility of distributed products?
- Are three additional and successful aseptic media fill runs required and performed after a media fill failure, to assure the sterility of products?
- Are all process changes incorporated into media fills in a timely fashion?
- Do the media fill batch sizes correspond to those specified in the validation or qualification protocol?
- Do media fill records document interventions in such a way that each intervention can be linked to the person or persons performing the intervention?
*Prepared from recent 483 observations
To track inspection results from the rest of the world outside the European Union and the United States, you must visit each individual agency or inspectorate’s website and hope that at least some information is released (Table 1). For starters, try a live link to the Therapeutic Goods Administration (TGA), the Australian regulatory authority, which is listed on the EMA page for non-EU agencies. That link leads to a page with additional links to TGA inspection information as well as a link to the home page of the agency. On the home page, type a word such as “inspections” or a phrase such as “inspection reports” into the search box and start digging through the document links your search brings up. The information you want may be in there, somewhere (nobody said this would be easy). If not, try different search terms.
As an example, such a search turned up an interesting and pertinent 2011 presentation titled TGA’s GMP Audits: Trends and observations (www.tga.gov.au/pdf/presentations/presentation-omq-110912-norder.pdf). Alternatively, you can try a Google search using a phrase such as “tga australia inspections” (without the quotes) and mine the results, but this can be overwhelming if your time is limited. A recent search using this phrase turned up about 6,020,000 results! With experience, some cleverness, and skill, such a search can be refined, but there will probably remain hundreds or even thousands of links to explore.

Mining Information for Trends
The next step in setting up an inspectional RI system is to mine the data you have collected. The ways to do that are probably at least as numerous as compliance professionals, but two examples that have worked well are using pie-charted moving averages of GMP systems categories to get the big picture and using bar graphs of user-customized categories to get a closer look.
The six GMP systems categories used by the FDA are the quality system, the facilities and equipment system, the materials system, the production system, the packaging and labeling system, and the laboratory controls system. A discussion of these systems is beyond the scope of this article but can be found in the agency’s Guidance for Industry, Quality Systems Approach to Pharmaceutical CGMP Regulations. Likewise, the details of calculating moving averages is beyond the scope here, but fruitful discussions and tutorials can be found on the Web or by visiting the “moving average” Wikipedia page.
M
oving averages are generally recognized as useful tools for spotting trends and are finding increasing use in the biopharmaceutical and biologics industries. Figure 1 is an example of a pie chart created from 12-month moving averages of the six GMP systems.
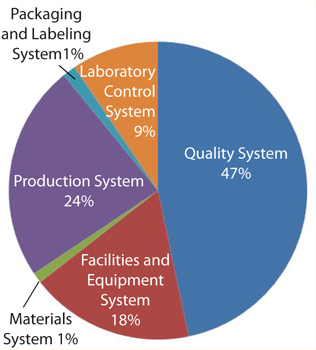
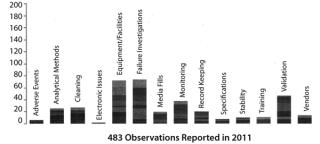
Figure 2 provides a better overview of leading generators of 483 observations. Deficient investigations lead the field and (unlike the data in Figure 1), equipment and facilities also loom large, most likely because I include observations about inadequate operational activities in this category. The advantage of this method is that each user can customize the categories tracked to fit the specific needs of his or her department or company.
Beyond Inspection Results: Advanced Trend Spotting
In addition to assembling and analyzing inspection results, other means of identifying and predicting regulatory trends include watching for regulatory cross-fertilization between agencies, frequent searches of regulatory authorities’ websites, nongovernmental websites, and relevant blogs, and (for the US FDA) keeping a close eye on what centers other than Center for DrugEvaluation and Research (CDER) and Center for Biologics Evaluation and Research (CBER) are up to.
Inspectors from many regions are now regularly sharing inspectional information through several means. Prominent among those is the Pharmaceutical Inspection Cooperation Scheme (PIC/S). All such activities are bound to result in the agencies incorporating what each conceives of as the others’ best habits into their own programs. So, if you see something happening in Europe, it is probably only a matter of time before the FDA picks up on it. In addition, the FDA, historically, has tried out many of its new initiatives in the Centers for Food Safety and Applied Nutrition (CFSAN) and Center for Devices and Radiological Health (CDRH) before bringing them to drugs and biologics. Recent examples of that include the initial rolling out of the Team Biologics Inspection program for medical devices and the present proposed trial of reinspection fees for food-processing companies.
For experienced regulatory professionals, enforcement and therefore inspection trends can often be inferred from study of Compliance Policy Guides (CPGs), which are FDA’s self instructions on the enforcement of its regulations.Especially relevant for readers will be the Biologics (Chapter 2) and Human Drugs (Chapter 4) sections. As a starting point, take a look at CPG Sec. 400.200 Consistent Application of CGMP Determinations, CPG Sec. 410.100 Finished Dosage Form Drug Products in Bulk Containers, Applications of Current Good Manufacturing Practice Regulations, and CPG Sec. 425.100 Computerized Drug Processing; and CGMP Applicability to Hardware and Software. Finally, a nongovernment website that may be of use is that of the European Compliance Academy www.gmp-compliance.org/eca_index.html). The FDA Law Blog (www.fdalawblog.net/fda_law_blog_hyman_phelps/) is also a fruitful source of information.
Putting Your Knowledge to Work
The final step is to put your knowledge to work by performing gap analyses and self-inspections from checklists and then following through with gap-filling and corrective and preventive activities. Those are made more efficient and effective when informed by — in addition to such bulwarks as regulations, guidance documents, and company policies — inspectional trends and specific types of observations or even the observations themselves. Careful analysis of the data you have gathered and mined will prove invaluable in creating your self-inspection and audit lists and in providing focus to your gap analyses and other compliance-enhancement initiatives and maintenance programs. The “20 Questions” box shows how a sample self inspection is one way your new information can be put to work for media-fill process simulations. In a future article, I will discuss top inspectional findings from various regulatory agencies and several recent European and US inspectional trends spotted using the methods described herein.
Author Details
Tom Pritchett, PhD, is the publisher of the weekly newsletter BioQuality and an industry consultant and trainer; biotech1ster@gmail.com.