“Learn from yesterday, live for today, hope for tomorrow. The important thing is not to stop questioning.”
—Albert Einstein
Single-use systems (SUSs) have been treated as novel technologies for some time. I have spent much of the past 10 years introducing clients to SUSs and integrating them into conventional processes. They are part of the biopharmaceutical development and production landscape and a mature, integrated option for bioprocessing.
The value of SUS integration is soundly substantiated: reduced cross-contamination risk in a multiproduct facility, reduced bioburden risk, overall cost effectiveness due to lower capital and cleaning labor, reduced cleaning, and timely integration. Effective integration into process and business systems is the difference between a successful application of a SUS and one that is not. Most of the industry’s focus has been on physical connections such as tubing, connectors, and other components. Very little focus has been on the business integration of such systems.
As bioprocesses are established and SUSs are integrated into operations, the focus moves to identifying parts, verifying compatibilities, writing specifications, approving drawings, and validating processes. End-user procurement/materials management groups become involved with inputting vendors into purchasing systems and finding current good manufacturing practice (CGMP) warehouse space for large volumes of CGMP-released SUS components. Usually, the “newness” of a process and a SUS consumes much attention, and very little time is spent on planning lifecycle management. As good as SUSs are, they are not perfect. Even their best applications are not without issues. Nonconformances (NCs), corrective actions/preventative actions (CAPAs), and changes occur with SUSs as they do with traditional processes and procedures.

Biological product life cycles are a familar concept. SUSs have life cycles and can change as well. Their changes can come from
- process improvements that address process shortcomings and adopt new technologies
- SUS components intended to fit operations better, address physical constraints, fit batch sizes, implement alternative vendors, and address vendor-initiated changes.
- subcomponents replaced with SUSs, when new materials are introduced, existing materials are modified, new technology is developed, different fabrication methods are applied, and new vendors and subvendors are added.
In such a life cycle, a SUS must meet all of CGMP requirements just like conventional technologies. As SUSs are adopted into CGMP operations, the process is expected to still be under control according to 21 CFR 210.1:
… the minimum current good manufacturing practice for methods to be used in, and the facilities or controls to be used for, the manufacture, processing, packing and holding of a drug to assure that such drug meets the requirements of the act as to safety, and has the identity and strength and meets the quality and purity characteristics that it purports or is represented to possess (1).
But what does control mean? One approach is to look at where we came from and “learn from yesterday.”
Learn from Yesterday
SUS have been in the industry for over 30 years. They were primarily made of polymers, and the two major systems were filters and silicone tubing.
Filters used membranes made of materials such as PTFE and cellulose acetate. They came first in cartridges and then in single-use capsules. Membrane and component manufacturing methods allowed for fully integrated systems. As those matured, filters were supplied with physical testing certificates to ensure their “filtration” performance.
Silicone tubing entered the biological market from the medical community. Doctors and hospitals went from using natural rubber to silicone. The tubing was initially supplied in rolls and with physical testing certificates to ensure its dimensions and “elastic” performance.
Two Worlds
Current single-use systems are hybrids
Raw Ingredient
- Lot control
- Mostly polymers
- Mostly vendor-controlled supply chain
- Validation controlled by vendors
Equipment
- Built to engineering drawings
- Sensors
- Larger volumes common up to 200 L
Those first single-use commodities were handled by biopharmaceutical companies (BPCs) as raw ingredients similar to media and reagents. Lots were controlled and tracked against specifications. Some companies performed quality control testing, but most relied on a “paper” approval and reviewed vendor-supplied certificates.
The supply chain was controlled by vendors before items arrived at BPCs, with many using a distributor network. BPC audits focused on basic control of components’ final form and very seldom looked into vendors’ supply chains. Those single-use components were then processed by BPCs, which constructed their own assemblies. All were configured for ease of use and to fit the configurations in the field — similar to what we ask vendors to do today. They usually were rinsed with water for injection, autoclaved and assigned a shelf life, used in processes, and discarded. More creative companies found ways to reuse some components.
Change References
21 CFR 211.100: Production and Process Controls, Written Procedures: Deviations
FDA Guidance for Industry Quality Systems Approach to Pharmaceutical CGMP Regulations
ICH Q7: Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients
Component lots were usually tracked in batch records for traceability, and the autoclave runs were validated. This worked very well for years and was primarily organized by manufacturing and quality groups. They are more familiar with raw ingredient specifications, quarantine and release procedures, manual equipment preparation, and what processes are required from a processibility and aseptic processing perspective.
Change management for those first single-use components was primarily through specification updates, driven by vendor changes or process requirement changes. Little to no testing was involved — perhaps an occasional cytotoxicity test, an audit, or autoclave run verification. Those first SUSs were either operated alone in laminar-flow hoods and biosafety cabinets or connected to validated process equipment. The validated systems were usually multiuse capital equipment, installed and validated as part of a one-time project and run for many years. Such equipment was primarily made of stainless steel, with a few polymers for seals.
The supply chain was handled exclusively by vendors. They primarily fabricated to drawings, made stainless steel to a specification, delivered equipment, and completed equipment “turnover” packages. Those packages out as files or binders of drawings, operating procedures, and instrument and fitting catalog
cuts. They have evolved to be encyclopedia-or terabyte-sized dossiers with material certifications, weld logs, instrumentation manuals, automation functional requirement specifications, and more. Turnover at the dock of a BPC was literal and symbolic. Except for some installation assistance, vendors were out of the picture. BPCs installed, checked-out, and validated systems with multiple use and maintenance considerations. Autoclaved filters, tubing, and sample SUS assemblies were integrated to equipment, with little to no vendor involvement.
Those processes were handled very differently than they were for the first SUSs and usually by different departments of a BPC (typically engineering, validation, and manufacturing). The usual links between the two types of process components — SUS and fixed equipment — were manufacturing and validation. In small operations, those are performed by one or two individuals to provide some continuity. In larger organizations, they are conducted by different groups and sometimes limited exposure to other types of equipment and their requirements. Change management for fixed equipment is by change control: a project-driven exercise with extensive testing, (requalification and revalidation) to verify that process requirements are met. All are performed by a BPC, with little or no involvement of the vendor.
Live for Today: Hybrid Technology
Current SUSs are hybrids (see “Two Worlds”). The levels of complexity are increasing as companies strive to do more with SUSs while simplifying and/or eliminating preparation and processing steps. Those steps are not going away, just being performed somewhere else by vendors. The key is to look at the total package and the value that it delivers; it is all contained in CGMPs.
Change management is handled differently for the two types of equipment. For raw ingredients, it involves specification change and ongoing testing or certificates of compliance to meet specifications, with involvement from vendors. Equipment change control involves specific project validation testing, then is allowed to run for normal operations with few checks and no vendor involvement.
Change management depends on your perspective and experience. I have been in meetings with BPCs and vendors at which the groups seemed to be speaking different languages. Let’s get back to basics and find some common ground between the two types of equipment. Regulatory documents (see sidebar “Change References”) contain the requirements for change management. Their interpretations have been diverse.
Effective change control activities (e.g., quality planning and control of revisions to specifications, process parameters, procedures) are key components to any quality system. For example, one FDA guidance discusses change in terms of creating a regulatory environment that encourages change toward continual improvement (2). So a manufacturer is empowered to make changes subject to the regulations based on the variability of materials used in manufacturing and process improvements resulting from knowledge gained during a product’s life cycle.
Why is change control so difficult? My father would say, “It is common sense!” What constitutes that, however, depends on who you are. Common sense to vendors focuses on components, subcomponents, and limitations in how they are made and distributed. Common sense to BPCs focuses on biopharmaceutical products and quality assurance. So it’s not simple. Although this “common sense” is in CGMPs, regulators have, in recent years, been telling us to consider risk. That is the issue!
Depending on where you sit, your perspectives on risk may differ. ICH Q9, CGMPs for the 21st Century, and ASTM E2500 are all useful guidances. Risk assessment is complicated between different individuals, not to mention different groups among multiple companies. SUSs typically involve at least four subcomponents from different vendors, plus an SUS vendor, plus end users, times multiple groups in each company. These interconnections can get very complicated (Figure 1). To help, changes in SUSs can be classified into four main categories, as follows.
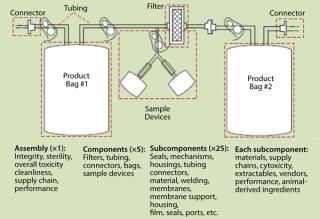
Material Change: Materials used in SUSs can be of complex construction. They are unique blends of base polymers, plasticizers, and fillers manipulated by proprietary processes to deliver amazing functions. Each has its own formulation and production process, which is usually the vendor’s intellectual property, and can be as complex as a final product. Raw material sources and processing methods change. Some are well defined, others (especially chemical substrates) are not. Their complexity alone makes them difficult to control, even if no changes happen. A raw ingredient or vendor change will heighten the complexity.
One risk is material interaction with a product and/or process. Vendors can evaluate that risk using biocompatibility and extractables tests. Most of the time, a BPC will perform its own leachable studies in addition to validation studies. Extractables and leachables testing has received much attention, and analytical methods have become more sophisticated. I see it similar to standards for water purity, which improved tremendously in the late 20th century. The risk is primarily toxicological, which is a well-defined science and needs a better basis.
Other risks may be present if an SUS uses a “polymer similar.” Vendors and end users are starting to conduct comparability studies to evaluate those risks.
Component configurations change when a vendor makes a change (e.g., to a manufacturing process or subcomponent). Product and process risks are usually related to process performance. For a vendor, the change can affect product performance (form, fit, and function). Sterilization assurance studies may be performed in such case. Questions to consider include whether a new component promotes more or less shedding. Will a new component or material increase risk to the process, thus increasing potential patient risk? Vendors that are more in tune perform their own risk assessments and present rationale that may include documentation of their own testing to support a change.
An SUS design change (e.g., size, process stream) is usually initiated by a BPC. The risk is also mostly to SUS performance: Does it meet process expectation? Does the process still work? When vendor changes occur, interconnections are a concern. How should vendors’ proprietary designs and their compatibility with other vendors’ components be handled? Some vendors have used conversion assemblies, which induces their own risks.
Medical Contamination
1800s Civil War: surgery on the field under a canvas, sanitization with alcohol, or even whiskey to control contamination of surgical instruments
Early 1900 World War I: wound irrigation and sulfa drugs introduced to treat wounds and address infection
1940s World War II: penicillin and Streptomycin applied to infections; sterilized serum IV bottles applied on the field
1950s Korean War: controlled environments for operating close to the front lines1960s-Vietnam: IV bags, an SUS cousin, introduced; medics able to ca
rry them in the field to treat patients
Late 1900s Hospital Use: nearly all patient treatments (e.g. IV bags, catheters, medical devices) as single-use applications
2000s Afghanistan: mobile hospitals with cleanrooms, full single-use including sensors in a battle field.
An SUS supply chain can include purchasing sources from actual SUS manufacturers, local distributors, local sales representatives, and large laboratory supply houses. This complex range directly affects the risk of control and tracking of an SUS. That risk is real and frequently overlooked, but addressing it is a CGMP requirement. Historically, companies received SUSs (filters and tubing), cleaned them, sterilized them, and ensured that they were controlled after processing. Today, companies hand those processes over to others farther up the supply chain.
The process can be made more difficult in cases with alternative suppliers. Even if all components are the same, their supply chains may be different, and mix-up risks can be introduced.
So how far upstream do we need to go with tracking the supply chain of subcomponents or polymer raw materials? How far do we go back? What testing is performed? Some tests are performed periodically or with every lot (e.g., safety assurance levels, endotoxin, and integrity). Others are performed on only the components or subcomponents, not an entire assembly. Some changes are significant enough to affect performance (basic function, extractables performance, or biocompatability performance). In such cases, tests can be significant in cost and effort, but they need to be considered with each change.
Do we need help, or is it all in the CGMPs? We need to agree on terms, and we need to listen to each other. I have been at vendor BCP meetings in which both sides are ready for battle. I help facilitate discussions and get both sides to share their views and open up to the other’s views. Unfortunately, there have been times when I served more as referee or a marriage counselor. The SUS gets implemented but usually with much difficulty. But when BCPs and vendors are partners, share responsibilities, and align the requirements, the process is much more successful, both initially and over the life cycle.
Hope for Tomorrow: Standardization or Not?
Many people are asking for standardization, but what does this mean? As discussed above, it depends on where you are coming from. Standardization is difficult in a changing industry, especially one such as SUSs (see the article by Maribel Rios on standardization in this issue).
The American Society of Mechanical Engineers (ASME) has existed for 125 years. It has issued standards for pressures vessels for that long, mainly driven by safety risks. Stainless steel equipment was used for biologics for many years before ASME’s Bioprocessing Equipment Standard (BPE) was introduced in the 1990s. It covers the design of equipment for sterility and cleanability to address contamination risk. The BPE evolved over time and today includes SUSs in addition to traditional stainless steel equipment. It will continue to evolve.
Standardization is problematic in the changing environment of SUSs. It will be good if it helps the industry apply new technologies for its betterment to incorporate new approaches, but conformance can also hurt growth and innovation. Two examples that come to mind are Apple’s iProducts and hospital applications of medical contamination risks.
The first Apple iPod was introduced just 10 years ago. The iPhone was introduced five years ago, the iPad two years ago, and Android devices three years ago. Could those devices have been possible if they had to comply with conventional computer standards at the time of their development? Obviously not. Can someone introduce an application today if it does not meet Apple’s or Android’s development guidelines? Obviously not. Where was the tipping point from “novel and new” to “standard”? The degree of standardization depends on where you are and when the market drives it. I have said at the BPE meetings, “We need the standard to allow for the next ‘iPhone’ in biopharmaceutical equipment when it comes out!”
The other example involves medical contamination risks. Surgical environments and medical treatments have evolved to improve survival rates from infection. We’ve witnessed real and effective microbial contamination risk reduction (see sidebar “Medical Contamination”). The level of microbial risk was reduced and will continue to lessen.
Organization Efforts
Consensus-driven groups are accredited with organizations such as ANSI and ISO.
ASME-BPE has a section for SUSs in its current revision, 2009. It will be enhanced in the 2012 revision, and SUSs will be an area of focus for the 2014 revision.
The Association for the Advancement of Medical Instrumentation (AAMI) has many initiatives that can satisfy SUS applications. AAMI is working on connector standards and balancing them against patient risk.
Industry-led organizations have diverse membership to align best practices.
A PDA Task Force is in the final stages of issuing its Single-Use Systems Technical Report, which should be issued in 2012. PDA will be holding a SUS workshop at its annual meeting. PDA’s supply chain references can be applied to SUSs.
The ISPE Disposables Community of Practice has initiated an effort to write a baseline guide for single-use systems. Active work should start in 2012. Supply chain references can be applied to SUSs.
BPSA is holding a forum in July 2012 to discuss the issues with SUSs.
The Extractables and Leachables Safety Information Exchange (ELSIE) is an information exchange of companies sharing toxicological information.
Current Standards: Without clear standards for the biopharmaceutical SUS industry, we have had to borrow from other industries.
The food industry started with the first polymer materials for seals, diaphragms, and hoses. 21 CFR 177 contains a list of polymers. It is part of 21 CFR Part 170 and 180, which list materials generally recognized as safe (GRAS). Part 21 CFR 179 even has information for irradiation of food packaging materials. However, 21 CFR 177 is only for legacy materials, so not all materials are listed there. In 1997, the FDA Modernization Act (FDAMA) amended Section 409 of the FD&C Act to establish a food contact notification process that allows for faster review of new food contact substances (FCS) that are food additives. The European Union has its regulation on materials and articles intended to come into contact with food, (EC) No. 1935/2004. It includes a section on “special requirements for active and intelligent materials.”
Those guidelines have historically been the requirement for seals, gaskets, and hoses used with traditional equipment. Although they have been overlooked and surpassed by SUSs, they present similar risks, if not more risks to both materials and supply chain. Food regulations provide a good start but are not intended for parenteral biopharmaceuticals.
From the medical device industry, the SUS industry has also borrowed some guidance such as USP Class VI and ISO 10993 for biocompatibility (which have been referenced for years) and ISO 11137, sterilization of healthcare products. Radiation is being referenced primarily with sterilization assurance. Medical devices also have the 510K process for tracking changes similar to what BPCs have for their product licenses. It is currently being reviewed, and some changes are being considered. Many SUS vendors and subvendors already follow 510K for their products that cross over to medical devices.
Companies have also applied drug product standards. Final vial and stopper requirements have been referenced for years, and drug product requirements such as USP 788 for particulates have been referenced. Filters comply with 211.72 requirem
ent so as not to release fibers into products (or filters are used to remove fibers).
So what does it mean for a component to be FDA compliant? Each type of product above is administered by different branches of the FDA (Center for Food Safety and Applied Nutrition’s Office of Food Additive Safety; Office of Medicinal Products and Tobacco and Center for Devices and Radiological Health; Center for Biologics Evaluation and Research; and Center for Drug Evaluation and Research, respectively). The desire again is to have some order and differentiate among benchmarks, standards, and regulations.
The SUS industry has developed a fairly good approach to extractables and leachables, led by the Bio-Process Systems Alliance (BPSA) (3). It is starting to address particulates and integrity. As with everything, the challenge is that we have more information these days, and the requirements borrowed from other industries are becoming less applicable.
Standards and codes are developed and written to presumably allow their content to withstand the rigors of jurisprudence, should the engineering integrity of their use be questioned in a court of law. A guideline, on the other hand, serves to bring together best practices from a broad range of sources, with regard to system design, validation, and commissioning.
The consensus process for developing standards/codes and guidelines is a very effective means of drawing on diverse experience and first-hand knowledge in the industry. The use and practice of those standards and guidelines promote quality, continuity and reform within the industry while reducing the cost and labor for users who would otherwise be required to develop much-needed requirements at their own expense.
By referencing standards, codes, and guidelines in proprietary specifications, process equipment and system design, and a project’s basis of design, a company can benefit from the experience, knowledge, and know-how of dedicated industry experts. Furthermore, standards and codes become legal documents when referenced in a project’s agreed-to documentation. Should your project find itself in such a litigious situation, an added comfort is to know that some of what you base your contractual requirements on has been rigorously debated and scrutinized, making that aspect of a legal argument much more defensible.
SUS standardization efforts are under way (see sidebar “Standardization Efforts), mostly on the technical side. There is limited such activity in business practices, however. Supply-chain management may be another type of operation we can borrow from other industries, even from our own drug product supply-chain processes.
One seal supplier shared with me his surprise at the relative low level of component requirements in our industry. His company had a much higher standard to meet for automobile clients than for most of BPCs he dealt with. This was mostly in supply chain and management of change, but also in technical performance. As I discussed above, the tendency is to focus on the introduction of a SUS. Lifecycle practices can be referenced to other industries. Concepts such as vendor integration and just-in-time manufacturing will become more common in discussions of CGMPs and evaluating risks to patients and products.
We need to be cautiously optimistic about where we are going, with and without standardization. This is basic human nature and up to all of us in the industry. To quote Einstein again, “We can’t solve problems by using the same kind of thinking we used when we created them.”
Author Details
James Dean Vogel is founder and director of The BioProcess Institute: jvogel@bioprocessinstitute.com.
REFERENCES