It is estimated that hundreds of new recombinant proteins and monoclonal antibodies (MAbs) enter preclinical and clinical development each year (1, 2). Concomitant global competition in biologics manufacturing has put immense pressure to shorten the time to market. Over the years, cells from various origins have been used for therapeutic protein production (2, 3,–5). One of the most economical choices is Escherichia coli, used to make proteins such as human insulin and growth hormone. But the bacteria have some serious shortcomings, such as an inability to perform posttranslational protein modifications (6). Mammalian cell lines such as Chinese hamster ovary (CHO) cells, baby hamster kidney (BHK) cells, or human fibrosarcoma cells have addressed necessary glycosylation in production of certain therapeutic proteins (6). For MAbs, CHO and NS0 cell lines are the most commonly used expression hosts (2, 4).
PRODUCT FOCUS: ANTIBODIES AND OTHER PROTEINS FROM MAMMALIAN CELLS
PROCESS FOCUS: PRODUCTION
WHO SHOULD READ: CELL LINE AND PROCESS DEVELOPMENT
KEYWORDS: CHO CELLS, SERUM-FREE MEDIA, DHFR, TRANSIENT TRANSFECTION, DISPOSABLES, CELL LINE ENGINEERING, MTX, AUTOMATION, CELL SORTING
LEVEL: BASIC TO INTERMEDIATE
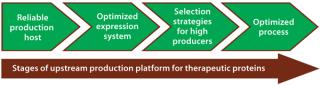
Usually for mammalian expression, stable clones with high specific productivities are the starting point for initiating development of a commercial biotherapeutic. This first step takes several months and can pose a major bottleneck in reaching the market. Since the early 1980s, advances in media development, selection strategies to identify highly productive clones, and nutrient feeding strategies have made it possible to get expression titers of 1–5 g/L in present-day bioprocesses (7). Figure 1 shows critical stages of an upstream production platform that relate to success of a therapeutic product.
With the alternative production platform of large-scale transient transfection, milligram to gram amounts of representative materials can be produced. That makes it very attractive for lead identification, initiation of preclinical studies, bioassay development, formulation development, and purification. By the time a robust clone can be generated and characterized, the necessary assays required to process commercial material would be developed. Here we discuss strategies that have significantly reduced biotherapeutic development time lines: expression vectors, clone screening, and clonal isolation methodologies, as well as alternative production platforms to rapidly generate milligram to gram amounts of protein.
Chemically Defined MediaCell culture media for mammalian cells have made significant advances since the development of tissue plasminogen activator (t-PA), which was the first FDA-approved biologic expressed by CHO cells (8). Originally, 2–20% serum was standard for growing adherent cells in vitro. But suspension cell cultures that could be rapidly scaled up are preferred for large-scale production. To substitute for serum in such cultures, companies initially used proteins such as albumin, transferrin, and insulin (9). But regulatory concerns over a high risk of adventitious agents directed media developers toward serum-free products.
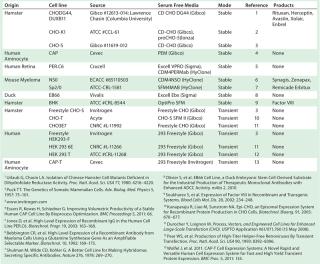
Chemically defined serum-free media (SFM) — with no animal-derived components — have become industry standard (10). High cell densities (>10 million cells/mL) and specific growth rates are routinely achieved using SFM. And most industrially relevant cell lines are preadapted to grow in SFM (Table 1) now, which reduces adaptation time (11). Invitrogen’s Gibco CHO-S cells were the first suspension-adapted CHO derivative available commercially. And cell-line–specific media are commercially manufactured for superior performance (Table 1).
Table 1: Industrially relevant cell lines suitable for rapid production of recombinant protein therapeutics in serum-free media (list is not exhaustive)
Almost all culture manipulations during cell-line generation — e.g., transfection, selection, limiting dilution cloning, cryopreservation, and recovery — are now optimized for serum-free conditions (12). Sometimes cells growing in suspension may aggregate or clump. Adding dextran sulphate or commercially available anticlumping agents to the culture media in optimal proportion can solve that problem. But such additives (as well as some other media components) are known to interfere with effective DNA delivery in host cells.
Commercial availability of serum-free and chemically defined media for widely used cell lines (e.g., CHO, NS0, BHK, HEK293) now allows researchers to readily establish their cell culture platforms. Chemically defined, animal-component–free (CDACF) media minimize batch variation and have simplified downstream processing by incorporating fewer contaminants, which helps in regulatory compliance. Modern, commercially available SFM boost cell productivity many fold over serum-containing media (12).
Customized for specific cell lines, such media support protein production rather than cell mass generation.
Although yields >20 g/L have been reported, cell lines are e
xpected to produce at least 1 g/L. To further improve product yield, a fed-batch process is usually chosen, with optimized supplementation of nutrient feed (13). Identifying the consumption rate of each nutrients by analyzing spent media usually gives a idea of cell-specific metabolic demands. Although nutritional demands of mammalian cell culture are widely accepted to be clone specific, bioprocessors would like generic media that could be used over a wide range of cell lines for commercial production. Considering the cost and time advantages of such platform culture media, vendors are working toward their development.
DHFR Amplification: In vitro construction of functional recombinant DNA (rDNA) paved way for expression of foreign proteins in host cells (14). After commercial production of insulin using bacteria, expression vectors were soon designed to produce recombinant proteins using mammalian cells (15). In the 1950s, investigation of cancerous cells for resistance to chemotherapy agents such as methotrexate (MTX) had led to discovery of dihydrofolate reductase (DHFR) gene-amplification mechanisms (16,–18). That formed the original basis of recombinant protein production in CHO cells without DHFR activity.
DHFR-deficient CHO host cells are transfected with an expression vector containing the DHFR gene under a weak sv40 promoter and the gene of interest (for expressing a MAb or other recombinant protein) under a strong cytomegalovirus (CMV) promoter. Increasing concentrations of the MTX selection agent stepwise (from 50 nM up to 5 µM) leads to several hundred copies of the DHFR gene (19). During that amplification process, a proportional increase in the copy number of a nearby linked gene of interest dramatically increases the clones’ specific productivity levels. An altered DHFR mouse cDNA differing in one amino acid has showed abnormally low affinity to MTX (20). That variant was used as a dominant selection marker for cultured cells that express normal levels of the DHFR enzyme (20).
Regardless of the DHFR variant, each amplification step takes three to four weeks of labor, making this a time-intensive process. To minimize timelines, one-step increase to very high MTX levels can produce a few surviving clones that express high levels of DHFR. Unfortunately, such developments can lead to undesirable altercations in DHFR function (18). Nonetheless, incremental MTX concentrations in multiple steps have been optimized considerably to achieve product yields of >1 g/L rapidly (21, 22).
The advantages of DHFR expression vectors outweigh their limitations. Now off patent, the DHFR expression system is still used to produce high-expressing cell lines in conjunction with improved genetic elements and cell-line development parameters (23). In 2010, CMC Biologics (www.cmcbio.com) showed that including novel codon deoptimization (least preferred for translation) DHFR sequences in its proprietary vectors increased their stringency of selection and provided for rapid protein production (24). Invitrogen’s OptiCHO expression system is based on DHFR expression vectors.
The GS System: Another well-known example of gene amplification is the glutamine synthetase (GS) expression system (8, 9, 25, 26). GS is a dominant selectable marker that can be used with GS-negative NS0 cells (25, 27) as well as CHO cells that contain an active endogenous GS gene (26, 28). The latter, however, require the GS enzyme inhibitor methionine sulfoximine (MSX). About 20–25 µM of MSX is usually sufficient to suppress endogenous GS activity so that only clones that express high enough levels to titrate out MSX inhibition will propagate. Selecting cells at levels of 100–1,000 µM of MSX could provide gene amplification. Increases of 4–10 copies of the GS gene in NS0 and 200 copies in CHO cells have been observed upon amplification (25).
In some cases, high specific productivities could be achieved without amplification. Coexpression of E1a, a transactivator of the CMV promoter, increased product yields 10-fold (26). Because GS-derived cell lines require no external glutamine supplementation, very low accumulation of toxic ammonia is observed in these systems. So far, five FDA-approved drugs have been commercialized using the GS system, including MedImmune’s Synagis antibody and Roche’s Zenapax antibody.
Cell-line instability still poses a challenge for the GS expression system, especially with CHO-derived clones after MSX removal (29). Modification of the endogenous CHO GS gene expression profile after MSX removal is considered to be the major factor in that effect. In July 2011, Lonza announced development of GS negative CHO cells using zinc finger nuclease gene knock out technology to address stability issues while promising speedy cell-line generation.
Other Systems: Cell-line productivity depends on transcriptional activity at the site of recombinant gene integration. Several genetic elements can be included in an expression vector for high specific productivities right at the transfected pool stage: scaffold/matrix attachment regions (S/MARs) from Selexis (www.selexis.com), ubiqutous chromatin opening elements (UCOE) from Millipore (www.millipore.com) and stabilizing and antirepressor (STAR) from Crucell (www.crucell.com). Reviewed extensively by Kwaks and Otte (30), such genetic elements improve transgene expression and clonal variability independent of mammalian type (31,–33). Without amplification, they can generate cell lines with specific productivities in the range of 15–75 pg/cell/day. A number of therapeutic products are being developed using these novel expression-vector technologies, and we hope to see their FDA approval in near future.
Site-Specific IntegrationRecombinant gene integration sites dictate not only the levels of transgene expression, but also long-term genetic stability. Hence, directing exogenous integration into expression-favoring genomic “addresses” of a host cell line provides for predictable protein production (34). Furthermore, a single copy of the recombinant gene integrated at a favorable genomic sites can express protein amounts that are comparable to multiple copies in amplified cell lines (35, 36). That offers potential advantages of characterization and implementation of site-specific integration approaches to the biotechnology industry.
Some efforts have been made in that direction using recombinase-mediated cassette exchange (RMCE) (37,–39), which was reviewed extensively by Wirth et al. in 2007 (40). Briefly, the genomic integration sites are first tagged with specific-recognition DNA sequences, then a gene of interest is flanked by matching tagged sequences targeted in presence of recombinase enzymes. Ideally, a master cell line can be generated in which expression properties of a reporter gene could be transferred to the gene of interest through RMCE-based site-specific insertion (37, 41,–43). However, specific
productivities of cell lines generated thus far have been only moderate. More compatible integration sites need to be identified.
A stable clone with high specific productivity is a nonnegotiable requirement for commercially viable production of therapeutic MAbs. Identifying desirable high producers in a transfected pool of cells poses a major bottleneck in cell-line development because a transfected pool is crowded with low producers, and high producers are rare. Spotting a high-producing clone is sometimes described as “finding a needle in a haystack.” The classical method for clone screening and isolation uses limiting-dilution screening in which roughly a single cell per well is deposited in microplates. A predetermined ratio of conditioned media is mixed with fresh medium to provide better growth conditions for each single cell to divide. This manual technique is time-consuming and inefficient. With a limited number of clones screened, there is always a possibility of missing the best candidate.
FACS Screening: Many high-throughput screening methods are being developed to reduce the time and increase the efficiency of identifying high producers (44). Improvements in throughput have been achieved using methods based on fluorescent-activated cell sorting (FACS), which is a flow cytometry technology proprietary to Becton Dickinson and Company (www.bdbiosciences.com) (45). In general, the specific productivity of a cell is estimated based on a fluorescent signal, and desired clones are efficiently separated from thousands of candidates in a very short time span. In 2003, Brezinsky et al. demonstrated the efficiency of FACS-based screening for rapidly enriching high-producing cells by staining transfected cells with fluorescent-tagged antibodies specific to a protein of interest (46). Without using MTX, the team increased productivity 25-fold; with MTX amplification, that increase was >120-fold. The positive correlation of cell-surface expression proteins with the specific productivity of a clone provided a simple and rapid means to develop high-expressing cell lines (47). But implementation of the method requires product-specific, fluorescent-tagged antibodies.
A few groups are using green fluorescent protein (GFP) for selectivity. GFP has been used as a second transcription unit (48) and fused in-frame with a selectable, amplifiable metallotheonine marker to generate MTGFP (49). In both cases, higher productivities were observed with the GFP-based FACS method than with traditional selection. Positive correlation of GFP signal and protein production rate allow for rapid generation of clones without product-specific, fluorescent-tagged antibodies.
In one study, the surface protein CD20 was expressed as a reporter, and FACS sorting helped rapidly and accurately generate the protein of interest (50). Not only did monitoring CD20 expression help identify high producers, but as CD20 levels decline over time, unstable clones can be removed during early development stages.
Although identifying desired clones based on monitoring another protein improved the efficiency of generating high-producing cell lines, it also placed additional stress on the cellular protein synthesis “machinery.” In another FACS-based approach, fluorescently labeled methotraxate was used as a marker for identifying high producers (51). Cells with the highest fluorescent (MTX) signal sorted out using FACS had a high number of DHFR copies, which correlated to high productivities. That method produced amplified CHO cells with high specific productivities within a few weeks.
Automation: Automated systems such as the Clonepix machine from Genetix (www.genetix.com) and the Cell Celector system from Aviso (www.aviso.com) perform high-throughput cell screening and product-secretion–based clone isolation. Transfected pools are enriched under selective pressure. As positive transfectants recover from that pressure, cells are plated at low density in a semisolid medium supplemented with a fluorescently tagged antibody that is specific to the secreted product. Positive clones grow as individual colonies and are identified based on a fluorescent halo around them.
The intensity of the fluorescent signal and the size of the colony determine product secretion and clonal growth rates. Using an optimized program, desired clones are picked and deposited onto 96-well plates using a robotic arm. Clones thus isolated have been shown to maintain their productivity rankings upon expansion (52). Hence, high producers are identified at the first stage of screening, which subsequently shortens the overall development timeline. However, clone characteristics change from one host and expression system to the next, so clone rankings should be verified in shake flasks. And most high-throughput screening methods require expensive equipment.
Transient TransfectionProduction of clinical-stage products by stable cell lines is currently prevalent because it offers high yields, consistent product quality, and regulatory familiarity. However, milligram to gram quantities of proteins are often required in early development. Because stable cell-line generation is time-consuming and resource-intensive, transient transfection in mammalian cells is frequently used at this stage instead (5, 53). The procedure requires formation of positively charged polyplexes of supercoiled plasmid DNA carrying a gene of interest in an appropriate medium, then subsequently added drop-wise to exponentially growing mammalian cells. Pham et al. reviewed five key aspects of achieving highly efficient transfection (5): cell line, expression vector, plasmid DNA quality, transfection vehicle, and culture medium.
Originally, HEK293 cells were the host of choice for transient gene expression (TGE). By comparison, CHO cells are more challenging for transient transfection, usually with lower yields (54,–56). Product quality differs between the two cell lines (57), and CHO is the preferred host for commercial protein production, so most companies would rather use the same expression platform across their developmental work. Major effort has been invested on improving TGE in CHO cells, and productivities ranging from 10 mg/L to >100 mg/L can now be achieved (5, 58).
Engineered cell lines and feed supplements have increased overall process yields to improve the existing potential of TGE technology. Canada’s National Research Council offers CHO-3E7 cells with pTT5 vectors for licensing through its Biotechnology Research Institute (www.nrc-cnrc.gc.ca) (59). ACYTE Biotech’s Epi-CHO expression system is another example (59). Florian Wurm’s group at the Swiss Federal Polytechnic Institute in Lausanne is using CHO DG44 cells with XLG vectors (60). And Rosser et al. adapted CHO-K1 in suspension and used them at ≤1-L scale to produce milligram quantities of protein (61). Cevec’s human-aminocyte–derived CAP-T cells express a large T antigen of simian virus (SV40) in another unique transient expression system (62). And Invitrogen’s CHO-S cells using the Freestylemax transfection reagent are also promising (63).
However, the relatively low-cost polyethylenimine (PEI) reagent is preferred for transient transfection at large scales. MAb quality obtained
from a transient transfection was found to be the same as that obtained from stably transfected CHO cell lines (55, 58, 64). But process yields varied by batch based on various factors — of which transfection efficiency and cell health are most important. To minimize the time spent for seed-train development, “super cell banks” could be considered, with 200 million or more cells per vial.
Stably transfected pool technology is gaining popularity (65). Lucas et al. first presented the utility of stable pools in generating milligrams of recombinant protein rapidly by using a distronic DHFR expression vector and MTX selection (66). Unlike large-scale TGE, the transfection of stable pools is performed at small scale, then positive transfectants are enriched for a period of two weeks before expanding them in selective media. Using proprietary expression vectors, Selexis has generated a stable transfected library of 250 IgG-producing pools of CHO cells (67). The highest antibody titer on day 10 of a fed-batch process exceeded 1 g/L, demonstrating the advantage of transfected stable CHO pools to generate large quantities of protein rapidly for preclinical studies.
Stable pools are scalable up to 200 L (68), generating gram quantities of representative material without using large amounts of plasmid DNA. Once established, stable pools can provide small amounts of material repeatedly without additional optimization. However, their productivities can be increased by repeated cell sortings and inclusion of UCOE cis-elements in an expression vector (69).
Stable pools can be conveniently cryopreserved, which provides an optional stop point in a development program. When they are required, cryopreserved stable pools can be revived in culture and screened for high-producing stable clones. These pools are stable for a limited period, so it is critical that high-producing clones be isolated during this time window. Jianxin ye et al. have also shown that product quality — in terms of glycan distribution — of proteins produced by stable pools is similar to those expressed by clonal cell lines (68).
BacMam TechnologyKnowing that baculoviruses can infect mammalian cells and express a cloned transgene makes this one of the most promising methods for recombinant protein production. An engineered baculovirus containing active mammalian transcriptional elements (Invitrogen’s BacMam system) has been shown to express high levels of recombinant proteins in a range of mammalian cell lines, including HEK293, CHO, HepG2, and BHK (69). Unlike mammalian cell viruses, baculoviruses cannot replicate in mammalian cells — with inherent safety, negligible cytotoxicity, larger insert size, and easy scale-up. They are not pathogenic to mammalian cells, which makes baculoviruses an attractive tool for transient expression of proteins in mammalian cells.
Researchers from GlaxoSmithKline have implemented BacMam technology into pharmacologically relevant assays for drug discovery across its research and development program (70). Others have successfully applied BacMam technology for screening preferred drug targets such as G-protein–coupled receptors (GPCRs) (71), ion channels (72, 73), transporters (74, 75), nuclear receptors (76), and viral targets (77, 78). Moreover, baculoviruses have been used to deliver small interfering RNA (siRNA) into mammalian cells (79). Although some authors have highlighted the use of the BacMam system for recombinant protein production (80), little work has been done to expand the application of baculovirus transduction in this regard (81,–83). Nevertheless, the technology promises an efficient production system that could reduce time and cost in biologics development.
Disposable BioreactorsBecause of global competition and rising costs in biomanufacturing using cultured mammalian cells, some biopharmaceutical companies are implementing single-use technologies for small- to medium-scale production. Use of disposable bags for media storage, process development, seed inocula, and actual bioreactor vessels seems to be a solid trend. A major advantage of disposable bioreactors over stainless steel ones come in quicker turn-around times (with no steam sterilization or cleaning needed), which substantially decreases overall timelines and operational complexity.
Singh et al. introduced the first widely accepted single-use culture system in 1999: WAVE bioreactors (84). Mounted on a rocking platform, presterilzed bags can sustain a cell density of >10 million cells/mL in recombinant protein production. Without controls of dissolved oxygen (DO) or pH, this system’s main application is in preparation of seed inocula for large, stainless-steel, stirred-tank bioreactors. Wave-motion bioreactors are also preferred in generating bulk material with large-scale transient transfections.
Stirred-bag systems (e.g., the HyClone SUB single-use bioreactor from Thermo Fisher, the BIOSTAT CultiBag STR system from Sartorius Stedim Biotech, and the XDR-DSTB system from Xcellerex) are seeing more frequent use now, mainly due to broad experience already obtained with established stirred bioreactors (85). The Xcellerex bioreactors come in ≤2,000-L scale, and the HyClone SUB can accommodate 1,000-L bags. Ready-to-use, flexible polyethylene bags are fixed and shaped by a temperature-controlled stainless-steel container that is open at the top. An integrated, pitched-blade impeller with an angled shaft connected at the top (or a magnetically driven impeller without any shaft penetration) is used for culture agitation.
For successful implementation of single-use technology as a viable substitute for traditional stainless steel, stirred-tank bioreactors, it will be necessary to establish a broad range of reliable scale-up. Parameters such as mixing time and agitation rate especially need to be determined for every scale of working volume. Reusable stainless-steel bioreactors are characterized in terms of performance, optimization steps, and design. They have been the benchmark for many decades and are supported by a large amount of knowledge and process control experience among operators (86).
One study demonstrated that for high-density cell culture processes using CHO or NS0 cells, product from disposable stirred-tank bioreactors product from disposable stirred-tank bioreactors ≤1,000-L scale in a standard configuration was comparable with product from a 1,000-L stainless-steel bioreactor (87). The seed train, harvest, and downstream processing were performed using identical equipment and methods, so the only difference was the type of bioreactor involved. Encouraging results from similar comparable studies would provide the necessary impetus for full-fledged incorporation of disposable bioreactors into more biopharmaceutical facilities (62). The stirred-bag version has already been tested for potential application in continuous perfusion processes (88).
Speeding Products to MarketInnovations in mammalian cell culture, expression vectors, and the advent of automated technologies for clone isolation have been instrumental in drastically reducing development timelines for biologics. Large-scale transient transfection platforms are further assisting in expedited product development by quickly generating representativ
e materials. Every developmental method or tool poses its own advantages and limitations. So the choice of approach becomes critical to the success of a developmental program. Disposable bioreactors are gaining tremendous popularity among new and established biologics manufacturers. Growth and state of the art in the biotechnology industry promises to shrink developmental workflows by substituting time-intensive steps with rapid ones, thus shortening product and process development timelines.
Author Details
Corresponding author Vishal Agrawal, PhD, is a scientist in the bioproduction group at Aragen Bioscience; 380 Woodview Avenue, Morgan Hill, CA 95037; 1-408-779-1700, fax 1-408-779-1711;
REFERENCES
23:756-762.
-Up of Cell Cultures: The Next Level in Disposable Bioreactor Design. BioProcess Int 9:56-62.