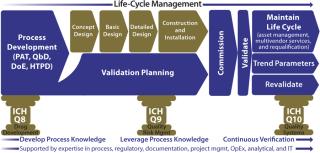
Biopharmaceutical manufacturers are striving to maintain productivity and profits while controlling increasing costs. Historically, validation has been seen as an expensive, non–value-added necessity to gaining regulatory approval to manufacture. Less often is it seen as a key element of an overall quality management system (QMS) that supports the safety, quality, and efficacy of end products for patients while also providing invaluable knowledge and experience for enhanced process control and management. When fully integrated with a QMS, a modular validation platform can offer biopharmaceutical companies considerable benefits:
-
cost containment
-
elimination of duplicate efforts and documentation
-
minimized impact of site changes
-
facilitation of effective and timely document approval
-
generation of a single test script detail for protocol execution record sheets
-
optimized record sheet preapproval
-
elimination of execution discrepancies
-
standardization of document formats
-
reduced document review and approval times
-
improved likelihood of on-time validation project completion
-
continuity of routine operations and life-cycle management
-
minimized exposure during regulatory inspections
-
assurance of regulatory compliance
-
proven history of regulatory success and user satisfaction.

Regulatory compliance can be a veritable mine-field for biopharmaceutical manufacturers. Facilities and processes that are “validated” to an inappropriate standard inevitably incur the wrath of the regulatory reviewers. Withdrawal of a license can result in loss of credibility and incalculable costs due to necessary remedial work, lost sales, and lost customers. All that can be easily prevented by adopting an appropriate approach to compliance from the start.
Planning, design, construction, commissioning, and qualification of a new or remodeled facility are substantial undertakings that often exceed their anticipated budgets. With increasingly stringent global regulations, rising manufacturing costs, and shrinking product pipelines, it is imperative that manufacturers achieve regulatory compliance for their production facilities, laboratories, systems, and processes more efficiently and effectively than ever before to remain competitive.
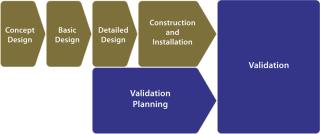
Facility validation programs are currently based on traditional complex models, in which validation planning and execution are often not even considered until relatively late in the project life-cycle (Figure 1). Lacking early adoption of an agreed-upon validation strategy, project cost overruns are inevitable, and facilities do not go on-line as scheduled. With the onset of validation planning, a level of inflexibility appears because of decisions already taken during earlier engineering process stages.
Most capital expenditure projects tend to be turn-key contracts to engineering, procurement, and construction companies, which also provide validation services. However, such companies often have limited knowledge outside their design, installation, and operation qualification (DQ/IQ/OQ) comfort zone. They rarely interlock their detailed designs and critical process parameters with critical quality attributes (CQAs) defined in product development. And they seldom get involved beyond OQ, at which point they generally expect a customer to take over the difficult work of turning qualification into validation.
That approach is normally mirrored by a highly detailed validation master plan (VMP) that focuses on elements of the project that the facility provider knows most about. This includes the facilities, utilities, automated systems, and equipment to be qualified together with documentation systems to be adopted in their qualification, validation, and use (protocols, raw data record sheets, reports, and standard operating procedures, SOPs). Obligatory product and process descriptions are also included, but simply in a way to justify the focus of the VMP. A token commitment is usually made to the ongoing operations of the facility by inclusion of a section on the types of SOPs to be generated and the maintenance, calibration, and training programs to be embarked upon.
On the surface, all that may look well conceived. But there is no upstream information from process development or downstream involvement by those responsible for the facility’s ongoing validated status. To an untrained eye, it might appear to fulfill the task. But to a regulator focusing on risk-based compliance, there is no guarantee that a product’s CQAs are being addressed by the process parameters being qualified — or that routine hardware maintenance will not lose sight of critical process parameters (CPPs).
Once such an approach to validation has been adopted, it is difficult to compile, control, and maintain. The effect of such a top-heavy strategy is that the validation phase of a project will often suffer from a requirement to update, modify, and have documents regularly reviewed and approved, which can lead to time delays and missed project deadlines. Resulting significant delays in facility completion can slow product manufacture, increase costs, and lessen revenue. Because process development is not linked to process understanding, validation decisions won’t necessarily be based on a product’s CQAs. That can lead to unnecessary validation efforts and/or cause important elements of a validation study to be missed.
Manufacturing sites that have been operating for a number of years often become focused on customer supply chain demands, which can lead to highly activity-based fast-tracking of new installations. In addition, start-up of new equipment, products, and associated processes can occur without the necessary diligence. With a broad portfolio of products, a high-activity culture, and the pressures of keeping up with product demand, it is easy to end up with a validation process built on weighty documentation packages that are autonomous and don’t support integration into a site’s overall quality management system (QMS).
A New ParadigmA solution that addresses the traditional shortcomings of validation is to introduce a modular approach that minimizes the impact of change and exposure of surplus information while optimizing the level of control and ease of regulatory/self inspection (Figure 2). Note that terminology varies across the industry, so the titles of documents and names of activities referred to in this article should not be assigned undue significance. Their satisfactory completion in the correct sequence is most important.
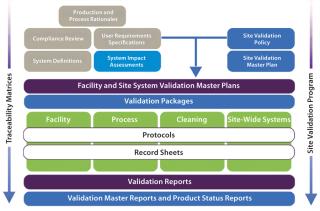
A modular validation platform is applied to all hardware and software systems used in a manufacturing process categorized as CGXP (affecting product quality). However, a number of critical project-based activities must be undertaken before such categorizations can be assigned. From a regulatory perspective, these are not a formal requirement, but they are nonetheless critical to the success of a validation project.
The Foundation: The justification to undertake a validation activity is derived from supporting evidence from activities shown in the top left of Figure 2: process and production rationales, user requirement specifications, compliance reviews, system definitions, and system impact assessments. Evidence-based deliverables determined by this process are mapped through to activity completion using traceability matrices. Figure 3 shows a schematic representation of our proposed validation process.
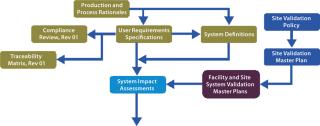
With this approach, production and process rationales drive validation efforts and provide clarity on what does and does not need to be validated. Dividing a facility’s validation process into logical chunks prevents unnecessary work. A process rationale that evaluates risks to product quality and formally documents CPPs is the starting point for a modular validation program. It forms a platform for a series of risk assessments performed while a user requirement specification (URS) and details of a full-scale production model are being developed. That provides a step-by-step analysis of a proposed production process and all the parameters that may affect finished product quality. It will also identify noncritical parameters and justify their lack of criticality. Major components of the modular validation approach are described below.
User requirement specifications are crucially important, holding all known requirements of all stakeholders. URSs are prepared before purchase of facilities, utilities, equipment, and operating systems. Within a modular validation project, nondetailed and flexible URSs act as a “master control” over more detailed system URSs. Requirements that affect product quality are automatically extracted from process and production rationales and compliance information from a regulatory database. Although there will be enough information for prospective vendors to satisfy all necessary requirements, URSs are flexible enough to allow cost-effective and compliant solutions to be put forward. Because validation protocols import their acceptance criteria from URSs, a site project or operational change management system ensures that all modifications are recorded and the validation program updated accordingly.
With this particular style of URS, additional economies are achieved in the build of later documentation. Aspects of a product’s life-cycle and parameters critical to product quality will be automatically inserted from process and production rationales, with associated regulatory requirements extracted from a database of regulatory information. This minimizes the risk of noncompliance, removes transcription and interpretation errors, and obviates the need to search manually through regulations and guidance documents.
System Definitions: Another key component of modular validation is a system definition document, which identifies the physical boundaries of each system and describes its complete set of attributes. Prepared before a system impact assessment, this enables descriptions in validation plans to be minimized but supported by cross-referencing. For example, a complex, automated clean-in-place (CIP) system linked to a number of other systems would have a precise and information-heavy system definition, whereas for a simple pH meter, a vendor brochure may suffice. System definitions eliminate the recreation of system descriptions in later, subservient, documents. They also remove the potential for loss of critical process and equipment information due to tailoring of specific descriptions for the introductory sections of protocols, and so on.
Validation Plans: As discussed above, validation plans (facility, manufacturing, and cleaning) for a site need not be detailed and inflexible documents. Their system descriptions can simply be cross-referenced to system definition documents, which allows changes (other than to critical parameters) without plan revisions. Essentially, the VMPs and validation plans serve as high-level documents that refer to more specific and appropriate documents. This approach helps minimize the impact of site changes, each of which can be recorded and documented in the most appropriate plan.
For a new or existing facility, it is important that the level of detail in VPs/VMPs is suitable to the knowledge and information available at the time they are generated and then detailed as the validation project develops. This saves time and money because plans won’t need to be rewritten, revised, and reissued. With most content as boilerplate text, author involvement is minimized, and the opportunity for human error is significantly reduced, saving department-head review and approval time.
Protocols and reports also benefit from a modular approach because they are developed such that each series of prerequisites, checks, and tests has its own objective, methodology, and acceptance criteria selection. Details (e.g., test scripts) and record sheets are separated from protocols to reduce review and approval times.
Automated Documentation: User-friendly automated templates are an important facet of the modular approach that allows authors to select appropriate documents from a menu that includes site validation policy, traceability matrix, and compliance review. Software then guides users to the correct part of the document, and changes/additions are automatically fed consistently throughout it where required. Each template also contains all the relevant and latest acceptance criteria to fulfill FDA and EMEA requirements. Automated reports for discrepancy-free executions can be generated in just a few minutes, and even outstanding or resolved discrepancies would be recorded with minimal author involvement.
Integrated StructureThe highly structured design of a modular validation strategy (Figure 2) provides a fully integrated process that supports full transparency of information on a number of levels, from plans and protocols to reporting and regulatory audits.
Each functional section of the overall validation strategy commences with either a plan or protocol (e.g., a facility validation plan, FVP). Completion of each stage is verified with a report, thus ensuring that all validation activities are closed out logically. (For example, a facility validation status report (FVSR) closes out against initial planned activity in the FVP.) This makes it quick and easy to access information at all levels of a site validation process and meets requirements for information to support assessment of change-control procedures or to answer specific questions during an audit.
Key reporting levels support the instant availability of status information that is required to support annual product reviews (APRs) and product quality reviews (PQRs), removing the annual event of building ad hoc reports to capture what validation activities have been completed during the preceding twelve months. Such information will already be available in documents such as a facility validation master report (FVMR). Once the FVMR has been approved, it can be used by QA/QC to support APR/PQR processes without further specific input from validation team members (a more efficient use of resources).
Regulatory audits may be either process and equipment based or product specific. Modular validation reporting structure supports either direction. A site validation master report (SVMR) is available to support process and equipment audits, and a product status report (PSR) supports product-based audits.
Future PerspectivesWith increasing industry costs and competitive pressures, a changing regulatory environment, and the availability of improved approaches to validation and document management, now is an ideal time for biopharmaceutical manufacturers to review their facility validation procedures and policies to establish more effective platforms for future operations. We believe that traditional approaches are often too complex as well as time- and cost-intensive. Our research shows that implementing a modular approach will help shorten the time it takes to bring a facility on-line and produce better quality medicines faster than before by eliminating unnecessary validation work while ensuring that necessary work is performed and documented correctly and accurately. Effective validation ensures better commissioning, good engineering practices, higher documentation standards, a platform and reference for future validation programs, better employee appreciation of the process, and ultimately a better guarantee of success.
Looking to the future, the industry is currently discussing “risk-based” validations, and regulatory authorities are certainly advocating the use of such an approach 2,. This is the first step toward total integration of validation activities into an overall QMS approach to continuous verification. Our industry, however, is still a long way from fully realizing this vision.
Inevitably, the biopharmaceutical industry will develop strategies to support risk-based approaches for manufacturing and validation. We are already seeing early process development become more important by leveraging high-throughput techniques to expand process understanding and enable process analytical technology (PAT). And to drive document generation and project management, more automation is being considered. These approaches will allow biopharmaceutical manufacturers to initiate validation efforts early and to validate correctly.
This current version of the modular validation platform supports a smooth transition to a desired future state. It maintains currently understood terminology while driving toward the new direction — by, for example linking to product knowledge very early in a project life-cycle. Because of its modularity, this process facilitates integration into ongoing operations by, among other things, the reexecution of particular validation package elements deemed critical to product quality as part of a routine, ongoing maintenance cycle. In addition to serving as a springboard toward continuous verification, the overall platform will become even more robust with inclusion of other stages of prevalidation work including factory acceptance testing (FAT), commissioning, and site acceptance activities. All of those can benefit from fully automated transfer of information from specification documentation straight into test document sets (Figure 4).