
Antibody-based drugs represent a growing segment of the pharmaceutical industry and one of the most promising classes of therapeutic drugs. The demand for protein-based drugs is expected to grow steadily for the foreseeable future. However, production of monoclonal antibody (MAb)–based drugs remains very costly, and solutions to lower production costs are needed. Downstream purification steps are becoming a target for cost reduction. The use of ion-exchange chromatography to directly capture MAbs from cell culture streams or as a polishing step after an affinity separation may be a way to reduce costs.
Ion-exchange chromatography is an established step in the purification of MAbs following protein A capture. In this application note, we demonstrate the utility of the combination of a cation exchange step and an anion-exchange step using UNOsphere Rapid S media and UNOsphere Q media (Bio-Rad Laboratories, Inc.), respectively. The mirrored binding characteristics of UNOsphere Rapid S and UNOsphere Q media provide complementary purification steps for the removal of host cell proteins (HCPs), fragmented or aggregated MAbs, leached protein A ligands, DNA, and virus particles (Figure 1).
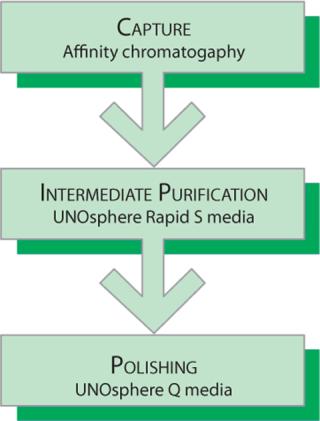
In the separation method described in this note, a protein A support was used to isolate a MAb from a cell culture feed. A large proportion of HCPs and other contaminants did not bind to the protein A support. However, along with the intact MAb, variant forms were retained by the protein A support and found in the eluted fractions. We used UNOsphere Rapid S media to separate a deamidated degradation product from the intact MAb and used UNOsphere Q media to further reduce the remaining contaminants to acceptable levels.
Materials and MethodsMAb was captured from a cell culture filtrate by affinity chromatography using immobilized protein A (0.5 × 5 cm column), equilibrated with 50 mM sodium phosphate, 100 mM sodium chloride, pH 7.0. Bound MAb was eluted with 100 mM arginine, 50 mM sodium acetate, pH 3.8. The MAb pool from protein A capture was adjusted to pH 4.5 prior to cation exchange chromatography.
For cation exchange chromatography, a column (0.5 × 5 cm) was packed with UNOsphere Rapid S media and equilibrated in 10 mM sodium acetate, pH 4.5 (buffer 1). An aliquot of the protein A eluate was diluted 1:4 with buffer 1 and applied to the column at a linear velocity of 300 cm/hr. The column was washed with buffer 1 and then with buffer 2 (10 mM sodium citrate, 10 mM sodium phosphate, pH 5). Elution was performed with a 15-column-volume pH gradient (pH 5–7.4) formed between buffer 2 and buffer 3 (buffer 2 adjusted to pH 7.4).
The main peak of MAb eluted from UNOsphere Rapid S media (8.0 mL) was adjusted to pH 7.0 by addition of 0.8 mL of 500 mM sodium phosphate, pH 7.0, and applied at a linear velocity of 300 cm/hr to a 0.5 × 5 cm column packed with UNOsphere Q media. In this flow-through step, the column was equilibrated with buffer 4 (50 mM sodium phosphate, pH 7.0). Following sample application, the column was washed with 30 mL of buffer 5 (75 mM sodium phosphate, pH 7.0) and then was washed with buffer 6 (500 mM sodium phosphate, pH 7.0) to strip the column of remaining bound components.
Protein fractions were analyzed by SDS-PAGE using a Criterion™ Tris-HCl 4–20% gradient gel (Bio-Rad). Mass spectrometry analysis of gel-purified proteins was carried out with an Autoflex II MALDI-TOF mass spectrometer (Bruker Corporation).
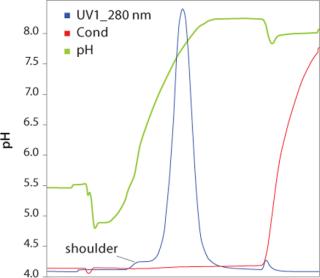
ResultsThe UNOsphere Rapid S media cation exchange chromatogram is shown in Figure 2. The bulk of the MAb eluted as a sharp peak during pH gradient elution. Recovery of MAb in the main peak, estimated by A280 nm, was >95%. Mass spectral data demonstrated a peak at 2138.6 amu (atomic mass unit) for the eluate pool and 2136.1 amu for the leading shoulder. This is consistent with deamidation of three aspargine or glutamine residues of the MAb. Such a deamidation event, which converts neutral asparagine, glutamine, or both to their respective acidic forms, would be expected to result in a more acidic protein that would elute earlier than the unmodified MAb in a pH gradient. With a shallower pH gradient than the one employed here, baseline separation of such species is possible.
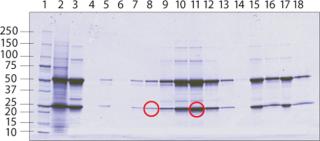
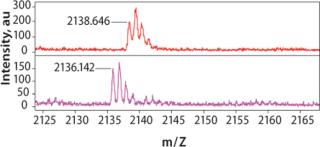
Aliquots from different fractions collected throughout the entire procedure were analyzed by SDS-PAGE (Figure 3). IgG heavy and light chains were present not only in the main elution peak, but also in the leading shoulder. Identification of the leading shoulder of the peak as a modified or fragmented MAb by gel electrophoresis was not conclusive. Protein bands from UNOsphere Rapid S media elution fractions 1 and 2 (shoulder), and 3–6 (main peak) were extracted from the gel and analyzed by mass spectrometry (Figure 4). Comparison of the measured mass of the shoulder fraction proteins and main peak proteins is consistent with three deamidations on a single MAb peptide.
The reduction of HCPs is shown in Table 1. HCP reduction was determined using ELISA quantification. UNOsphere Rapid S media chromatography achieved a tenfold reduction in HCP, and the UNOsphere Q media step further reduced the HCPs to below the limit of detection of the assay.
Table 1. Purification process. HCP content after each step of the MAb purification procedure was determined using standard ELISA techniques.
UNOsphere Rapid S media chromatography with an appropriate pH gradient elution scheme can be used to remove unwanted degradation products from a process stream with nearly quantitative recovery of IgG. In addition to providing a powerful separation technique, pH-based elution is well-suited to a process flow. Use of pH rather than ionic strength for elution means that the eluate pool will have low conductivity; the pool can thus be applied directly to a subsequent anion exchange step, if desired, with minimal feed stream dilution.