In 1982, authentic human insulin produced in genetically engineered Escherichia coli (under the trade name Humulin, from Genentech) was the first recombinant protein to be licensed by regulatory authorities for therapeutic use. That licensure and the subsequent production of human growth hormone heralded a new era of biopharmaceuticals and were the first examples of the industry's future potential. Both proteins are produced as fully active biologicals in E. coli. Insulin and human growth hormone are fairly simple small proteins, without complexities such as posttranslational modifications, which are required for many bioactive glycoproteins.
E. coli bacteria are ideally suited for producing small proteins, primarily because of their extensive genetic and metabolic characterization, fast growth rate, fermentation ability at very high cell densities, and relatively low costs associated with large-scale production. However, bacteria lack the genetic, metabolic, and posttranslational mechanisms required for faithful expression of high– molecular-weight glycoproteins (e.g., antibodies), which make up the bulk of emerging immunotherapeutic molecules. So alternative systems for expressing fully active glycoproteins are required to produce complex molecules at large scale.
Mammalian cell lines are premier expression platforms for producing complex biologicals. Chinese hamster ovary (CHO) cells have become the standard mammalian hosts for such proteins. Their success can be attributed to the cells’ extensive characterization, history of regulatory approval, fast growth rate, and adaptability to growth in serum-free medium (SFM) formulations in large stirred-tank bioreactors.
In the 1980s, researchers from Celltech Biologics introduced a murine myeloma cell line (B lineage) designated as NS0 (nonsecreting null) that was particularly well suited to produce immunotherapeutics. NS0 is a nonsecreting cell line and a glutamine and cholesterol auxotroph that does not produce immunoglobulins. So development of an expression system based on the glutamine synthetase (GS) gene as a metabolic selection marker effectively introduced NS0 cells to the biologicals industry.
In the early 2000s, Crucell introduced Per.C6, a human embryonic retinal cell line transformed with adenoviral sequences. This cell line offered several advantages over CHO and NS0 cells because of its human lineage origin, well characterized history, and higher regulatory compliance. One of the most prominent features of the Per.C6 line was its faithful glycosylation of human proteins, including antibodies.
Advancements
Immunogenicity: The structure of sialic acid residues that comprise the carbohydrate side chains on antibodies produced in mammalian cell platforms has long been a concern in immunotherapeutics development. Nonnative glycosylation patterns that decorate recombinant human proteins can render immunotherapeutics more immunogenic (less potent) and thus less effective. Several cases have been reported of such glycosylation aberrations in recombinant proteins produced in both CHO and NS0 cells.
New molecular biology technologies introduced from the 1980s through the 2010s have advanced the industry's quest to produce ever more faithful, potent, and effective immunotherapeutics. Such technologies include chimerization, humanization, affinity maturation, isotype and class switching, site-specific mutagenesis of recombinant antibodies, and expression of proteins fused to polypeptide partners aimed at increasing half-life and stability in vivo.
Animal Sera: To date, nearly all mammalian-cell–generated biopharmaceuticals approved for human therapeutic use are expressed from either CHO or NS0 host systems. Traditionally, the generation of stable, high-producing cell lines relies on medium formulations supplemented with animal sera, primarily of fetal bovine serum (FBS) origin. The high protein content of animal sera, however, poses a significant challenge in downstream purification of recombinant proteins. Thus researchers have developed robust SFM alternatives.
The advent of SFM for large-scale immunotherapeutics production has spawned a new and competitive industry for designing and implementing robust, chemically defined (CD), more regulatory compliant, and customizable formulations. In the late 1990s, researchers identified epidemic infections of livestock with transmissible spongiform encephalopathies (TSEs), namely bovine spongiform encephalopathy (BSE). The lack of reliable diagnostics for those agents meant that animal sera used to propagate cell lines could be contaminated with TSE. So many companies accelerated the development and marketing of robust CD media. Currently, robust CD media alternatives are available from a large number of medium manufacturers.
Acronyms
ADCF: Animal-derived-component free
BSE: Bovine spongiform encephalopathy
CD: Chemically defined
CHO: Chinese hamster ovary
DoE: Design of experiment
FBS: Fetal bovine serum
GS: Glutamine synthetase
Ig: Immunoglobulin
LDCC: Limiting dilution cell cloning
MAb: Monoclonal antibody
NEAA: Nonessential amino acids
NS0: Nonsecreting null
PF: Protein free
SFM: Serum-free medium
SPR: Specific productivity rate
TSE: Transmissible spongiform encephalopathy
Despite the availability of robust CD media, classical methods for generating stable mammalian cell lines still require serum-containing medium at the outset. Stable cell lines are grown and expanded in serum-containing medium until a selected few clones with high specific productivity rate (SPR) are chosen for SFM adaptation. Adaptation to SFM is performed stepwise or directly, and both methods have met significant success. The dual facet of the stable cell line generation process results in significantly extended production timelines and availability of products for in vivo studies (e.g., toxicology and clinical studies in human subjects).
Cholesterol Biosynthesis
The availability of robust CD media (specifically NS0 CD formulations) presents a unique opportunity to revolutionize the process for generating stable cell lines. One method takes advantage of cholesterol biosynthesis —another known metabolic auxotrophy of NS0 cells. Supplementation of exogenous cholesterol in NS0 SFM is critical for cell growth (1). Adding cholesterol to SFM is problematic because the lipid must be coupled to cyclodextrins to enhance its aqueous solubility. In addition, the supplementation of exogenous cholesterol into media and subsequent filtration results in losses that cannot be readily measured and quantitated. Such losses could significantly affect the optimal growth of cell lines.
NS0 SFM Expression System
Rescuing the cholesterol auxotrophy of NS0 with a single gene would potentiate the development of a novel expression system well suited to SFM applications. Selection medium would be supplemented with highly soluble glutamine but would lack cholesterol. The cells would synthesize cholesterol upon complementation of the enzyme required to rescue the deficient cholesterol biosynthesis pathway.
In attempting to design this novel expression system, I sought sought an enzyme with 3-ketosteroid reductase or dehydrogenase activity based on biochemical literature of the cholesterol biosynthesis pathway, specifically as it pertained to NS-lineage–derived cells. A plasmid bearing the murine 17 beta-hydroxysteroid dehydrogenase type VII (17β7) under the control of a minimal mammalian promoter effectively rescued cholesterol auxotrophy in NS0 cells selected in SFM.
Several observations added to the likelihood of successfully developing an NS0 SFM expression system. Parental cells are directly adaptable to basal CD media. After several passages, the cells displayed growth rates similar to those reported in serum-containing media. NS0 cells grow as loosely attached or as suspension cells in serum-containing media. Those cells retain the same morphology when adapted to SFM, with the added advantage of adopting enhanced suspension growth characteristics.
Transfectability and selectability of CD medium-adapted NS0 cells were subsequently assessed in media without serum supplementation. Whereas all commercial CD formulations support growth of high-density NS0 cultures, to date no single platform sustains cell growth at the low-seeding densities in posttransfection plating (usually in 96-well plates) required to achieve isolation of transfectants at near clonal levels. Further, limiting dilution cell cloning (LDCC) of NS0 lines is not possible in basal CD media. These formulations lack crucial components that permit the survival of single cells in a well (2).
Supplement: A rationally designed supplement was formulated and tested for its ability to adequately support the growth and selection of NS0 cells following transfection and seeding, as well as in LDCC. The supplement can be formulated either with or without CD components, thus supporting both research and production-grade platforms. The supplement is added to CD media and is used only during the initial selection stage. Upon expansion of cell isolates from 96-well plates to larger well plates, the supplement is no longer required. Likewise, clonal lines that emerge from LDCC do not require the supplement once the culture is transferred from 96-well plates to 24-, 12-, or six-well plates.
Cell Line Isolation: Finally, the combination of a plasmid bearing a 17β7 selectable marker transfected into CD medium-adapted parental NS0 cells, seeded in 96-well plates and selected in the same medium containing a serum-free growth supplement, permitted the isolation of stable cell lines. This system was next used to express human antibody genes cloned in a bicistronic vector — with light and heavy chains cloned in tandem or opposing orientations.
Results showed that SFM-adapted parental NS0 retains superior transfection properties, producing on average >3,000 isolates per experiment (10 million cells were transfected and plated per experiment) (Figure 1). This very high transfection and selection rate is 10× greater than the average obtained with serum-supplemented medium that supports emergence of NS0 lines using the GS selectable marker.
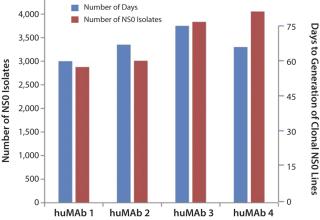
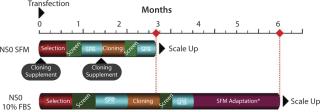
The combination of expression vector elements, medium platform, custom supplement, transfection, and selection methodologies have permitted generation of stable NS0 clonal cell lines in 60–75 days (Figure 1). This timeline comprises transfection, selection, screening, expansion, SPR assays, followed by LDCC, screening, expansion, and SPRs on clonal lines (Figure 2).
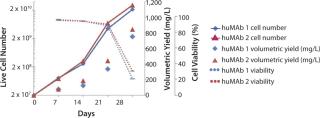
A number of stable NS0 lines have been generated using this system to produce a panel of immunotherapeutics based on human antibodies. Such therapies are under evaluation as countermeasures against viral hemorrhagic fever infections. We have generated several antibodies using this system, each requiring only expansion and culturing in 10-layer HYPERFlasks (from Corning) to achieve final yields of purified material at sub 10-g scale, with yields in the range of 750–800 mg/L (Figure 3).
Previous work demonstrated the feasibility of reverting the dependence of NS0 lines on exogenous cholesterol by stepwise adaptation to growth in medium lacking the lipid. Despite that, our work demonstrated that cholesterol auxotrophy in NS0 cells can be stably rescued with expression of an HSD17β7gene. To date all stable NS0 SFM clonal lines generated with the system described herein have retained expression and SPR levels for >22 generations (experimentally determined in production runs from cryopreserved cultures to scale-up in multiple HYPERFlasks).
These results demonstrate that methods used to generate stable NS0 SFM lines do not alter the characteristics commonly associated with this cell background — namely, stable and long term expression, fast growth and high productivity, and product consistency through multiple runs. Successful development of this expression system therefore depended on preservation of characteristics that make the NS0 cell background ideally suitable for production of large–scale biologicals, despite the need to overcome unique challenges.
The posttransfection and single-cell cloning steps present formidable obstacles to cell survival. Transfection results in greater than 99.99% cell death, as evidenced by 250–300 colonies per 10 million cells transfected using classical methods and selection medium. Thus surviving cells are exposed to significant pressure beyond normal selection because dying cells release harmful free radicals, proteases, and acidic contents of intracellular vesicles. This environment can contribute to the demise of otherwise stably transfected and viable cells. So it is important that cloning supplements be designed with such metabolic pressures in mind and that attention is given to components known to have protective properties.
It is not critical to formulate cloning supplements as protein-free (PF) or as animal–derived–component free (ADCF). If the supplement is designed as a component of a CD development process, it is, however, important that such proteins be of recombinant origin. Some proteins have critical roles in delivering essential metabolites to growing cells: quenching harmful free radicals in spent medium, providing protection from shear forces, and (coupled with other factors) even preventing the activation of apoptotic pathways and resulting cell death. Apoptosis is the preeminent mechanism by which cells die in culture. It can be induced by physical forces, a decrease in nutrients below a metabolic sustainable threshold, or an accumulation of harmful catabolites (e.g., lactate, ammonia, urea, CO2, and free radicals in spent media).
We achieved design and development of optimized NS0 cloning supplements through a design of experiment (DoE) factorial design, coupled with high throughput matrix analyses (4). The resulting supplements currently contain proteins as well as organic and inorganic compounds.
The cloning supplements developed are not clone specific, but rather cell-background specific. That result is yet another benefit of pursuing development of stable cell lines for production in SFM from the outset, because such supplements demonstrate a universal application within a particular cell background. This is demonstrated by the ability of the same cloning supplement formulation to sustain cloning of parental cells and individual stable transfectants with very similar efficiencies. Furthermore, the time to emergence of screenable clones or isolates in the original transfection when using SFM and cloning supplements is very similar to that in classical media containing FBS, (circa 2.5 weeks postseeding).
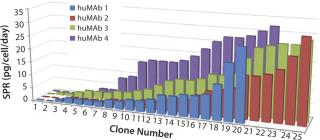
As stated above, cloning supplements are required to support only the initial growth of cells at clonal densities in SFM. When colonies reach cell numbers in the several hundreds or thousands per well, cultures can be expanded in SFM alone without detectable adverse effects. This rather rapid weaning process does not seem to affect the subsequent growth and production of cell lines through adaptation to scalable culture conditions, such as larger cell-culture plasticware (e.g., HYPERFlasks), spinner flasks, and stirred-tank bioreactors. So it appears that cloning supplements support essential functions critical for the survival of cells plated at clonal densities but do not affect long-term metabolic and functions of resulting cell lines. SPR analysis on the resulting pool of uncloned isolates generated with this novel expression system reveals a distribution of productivities similar to those obtained with classical methods — from zero to >30 pg of antibody/cell/day (Figure 4).
Development of supplements described herein that require limited use in low volumes and at reasonable costs will ensure not only efficient but also superior results in the generation of stable, clonal, production-quality NS0 cells lines. Although the development and implementation of an NS0 CD cloning supplement is currently feasible, the presence of specific protein factors from recombinant sources would make the cost per experiment expensive, although not prohibitive. Alternatives may need to be investigated, such as the attribution of CD designation to such components based on source or identification of other protein or nonprotein factors that would constitute acceptable substitutions.
Non-CD–designated protein components in formulating NS0 cloning supplements are an affordable and efficient alternative. If non-CD supplements are used to generate stable production cell lines, it could reduce development timelines and costs. But this method would not provide immediate advantages in regulatory compliance if one or more protein components originate from animal and/or nonrecombinant sources. In such case, adventitious agent and other testing requirements would not be necessarily reduced but would remain equivalent to those required with current methods used to generate stable production NS0 cell lines (Table 1). Regulatory concerns posed by the use of animal-derived protein components have, however, resulted in the development of ultralow endotoxin animal products specifically designed for SFM cell culture applications (5). Such products are made in current good manufacturing practice (CGMP) environments and are certified animal-virus free. They offer significant improvements in regulatory compliance over counterparts available until recently and are suitable for use as supplements in SFM formulations.
Table 1:
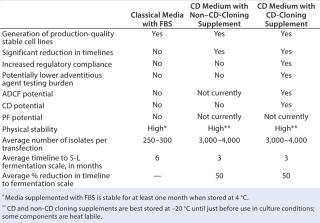
Several medium development companies have targeted the CD media niche markets by developing recombinant protein factors with crucial roles in mammalian cell cultures. They include recombinant forms of human insulin, albumin, and transferrin.
A High-Demand Market
To date, we have identified no disadvantages of adapting the methods described herein to generate stable NS0 cell lines to produce biological molecules using SFM from the onset of the process. Continued development and optimization of SFM — coupled with the use of CD or non-CD cloning supplements — offer an ideal platform for future generation of NS0 and other mammalian production cell lines (6). Advantages include significant improvements in critical parameters for sponsoring entities such as shorter timelines, enhanced regulatory compliance, significantly lower costs, and improved control of input medium components. With improvements in other biomanufacturing technologies (for recombinant protein glycosylation, high volumetric fermentation yields, and high-quality final products), such advancements are critical in an industry in which demand seemingly exceeded current manufacturing capacity by a factor of four in 2005 (7).
As a result of such demands, the biopharmaceutical industry continually strives to improve upon currently established cell line production and scale-up technologies to improve yields and reduce timelines and costs. The alternatives and methods described herein are steps toward achieving such goals in an industry forecasted to reach overall worldwide sales of $235 billion by 2015 (8). The global market for therapeutic monoclonal antibodies alone was estimated at $44.6 billion in 2011, and is expected to rise at a compound annual growth rate (CAGR) of 5.3% to nearly $58 billion in 2016 (8).
Author Details
Luis M. Branco, PhD is cofounder of Zalgen Labs, LLC Zalgen Labs, LLC, director of the Lassa Fever Program at Autoimmune Technologies, LLC (