 Antibody–drug conjugates (ADCs) use the targeting ability of a monoclonal antibody (MAb) to deliver a highly biologically active drug to diseased cells while sparing healthy cells, creating potent and effective therapies. This emerging class of novel drugs currently focuses almost exclusively on cancer treatment. Two blockbuster ADCs — brentuximab vedotin (Adcetris from Seattle Genetics) for treatment of rare lymphomas and ado-trastuzumab emtansine (Kadcyla from Genentech/ Roche, manufactured by Lonza) for treatment of HER2-positive metastatic breast cancer — have improved treatment options for many patients and constituted the majority of the US$454 million ADC market in 2013, according to the UK-based market research firm Roots Analysis (1). With more than 30 ADCs in clinical testing and many more in preclinical development, Roots Analysis estimates that by 2018 sales of ADCs will climb to $5 billion/year.
Antibody–drug conjugates (ADCs) use the targeting ability of a monoclonal antibody (MAb) to deliver a highly biologically active drug to diseased cells while sparing healthy cells, creating potent and effective therapies. This emerging class of novel drugs currently focuses almost exclusively on cancer treatment. Two blockbuster ADCs — brentuximab vedotin (Adcetris from Seattle Genetics) for treatment of rare lymphomas and ado-trastuzumab emtansine (Kadcyla from Genentech/ Roche, manufactured by Lonza) for treatment of HER2-positive metastatic breast cancer — have improved treatment options for many patients and constituted the majority of the US$454 million ADC market in 2013, according to the UK-based market research firm Roots Analysis (1). With more than 30 ADCs in clinical testing and many more in preclinical development, Roots Analysis estimates that by 2018 sales of ADCs will climb to $5 billion/year.
Although the basic principle seems straightforward, drug developers have had to overcome many challenges to generate safe and effective ADCs. The success of such products depends on four factors: the target antigen (with exclusive or increased expression on diseased cells), the targeting antibody, the linker, and the payload. Early linkers were either too stable (resulting in low potency) or too unstable, which led to high systemic toxicity. In fact, many early product failures are reportedly attributable to poor optimization. The first generation of ADCs faced many limitations such as
- limited expression of the target antigen
- immunogenicity of the MAb
- inefficient internalization of the conjugate
- insufficient potency of the attached cytotoxic payload.
Today’s approved ADCs have tackled many of those issues including antibody selection, cytotoxic payload potency, and improved antibody linkages. However, those products are heterogeneous mixtures of differing molecules. Within a single product, ADC molecules can vary in terms of both drug-to-antibody ratio (DAR) and conjugation sites. That variance creates significant therapeutic liabilities, with each distinct ADC in the resulting mixture having different pharmacokinetic, efficacy, and safety profiles.
Using clinically validated conjugation technologies, a drug payload is conjugated by a linker to accessible endogenous amino acids in an antibody: lysine or cysteine. Using those methods, the biopharmaceutical industry is learning that an optimal drug payload appears to be four drugs per antibody molecule. That assessment is based on the fact that too little conjugated drug results in an ineffective ADC, and too much can change the physicochemical properties of the product and cause significant problems with stability or quality (e.g., propensity to aggregate).
A typical antibody has about 80 lysines, about half of them on its surface and available for conjugation. For cysteine-based conjugation, the cysteines that comprise the four inter-chain disulfide bonds are targeted as conjugation sites, so ADCs can be generated with as many as eight conjugated drugs per antibody. Therefore, in either case, the conjugation reaction cannot be allowed to run to completion but is terminated once a DAR of 4 is reached. This expresses an average of the different ADC species present, however, with DAR ranges typically varying 0–9 to one. And even those species with a precise DAR of 4/1 include several combinations of available conjugation sites.
Inevitably, many of the species within a product mixture are likely to be inefficient in delivering the cytotoxic “warhead” into a tumor cell.
That may well explain why only an estimated 0.1–1.0% of the total systemic dose of cytotoxin ever reaches its target (2).
Building Better ADCs
Following on the success of that first generation, researchers are developing novel technologies to generate best-in-class ADCs to provide more effective therapeutics with less toxicity. Homogeneity is key to optimal drug design and product safety, so researchers are currently developing methods and technologies to reduce heterogeneity.
Site-Specific Conjugation: One way to generate ADCs with known DARs and predictable conjugation sites is site-specific conjugation. A Genentech study published in 2008 showed that site-specific conjugation of the drug payload improved the therapeutic index of ADCs (3). The authors engineered cysteine substitutions at positions on light and heavy chains that provided site-specific reactive thiol groups, and they conjugated a monomethyl auristatin E to an antibody against the ovarian cancer antigen MUC16. Results showed that the near-homogeneous composition retained the antitumor efficacy of heterogeneous ADCs while minimizing the systemic toxicity (3).
An elegant strategy for improving conjugation-site control is to use nonnatural amino acids (nnAAs). Peter Schultz’s research group at Scripps introduced nnAAs with novel structural, chemical, and physical properties that are not found in the 20 common amino acids (4). Reactive nnAAs can be incorporated into proteins, including antibodies, by engineering new cell lines for protein expression. An nnAA is incorporated at a defined and precise position using an amber stop codon, which usually would stop and truncate protein expression. A specific transfer ribonucleic acid (tRNA) that recognizes the stop codon and is charged specifically with the nnAA of choice causes continued translation and site-specific incorporation of the nnAA.
Researchers at Agensys and Ambrx have showed that site-specific, nnAA-conjugated Herceptin ADCs had a superior therapeutic window to ADCs using a cysteine-based conjugation method (5). The team compared a conventional conjugated anti-Her2 ADC with one against Her2/neu that was created using nnAA-based conjugation and showed that the nnAA-containing ADC was superior in terms of in vitro cytotoxicity, in vivo tumor efficacy, pharmacokinetic properties, and rat toxicology. They incorporated the nnAA para-acetyl phenylalanine at two unique sites on the antibody and covalently attached a cytotoxic drug to those sites.
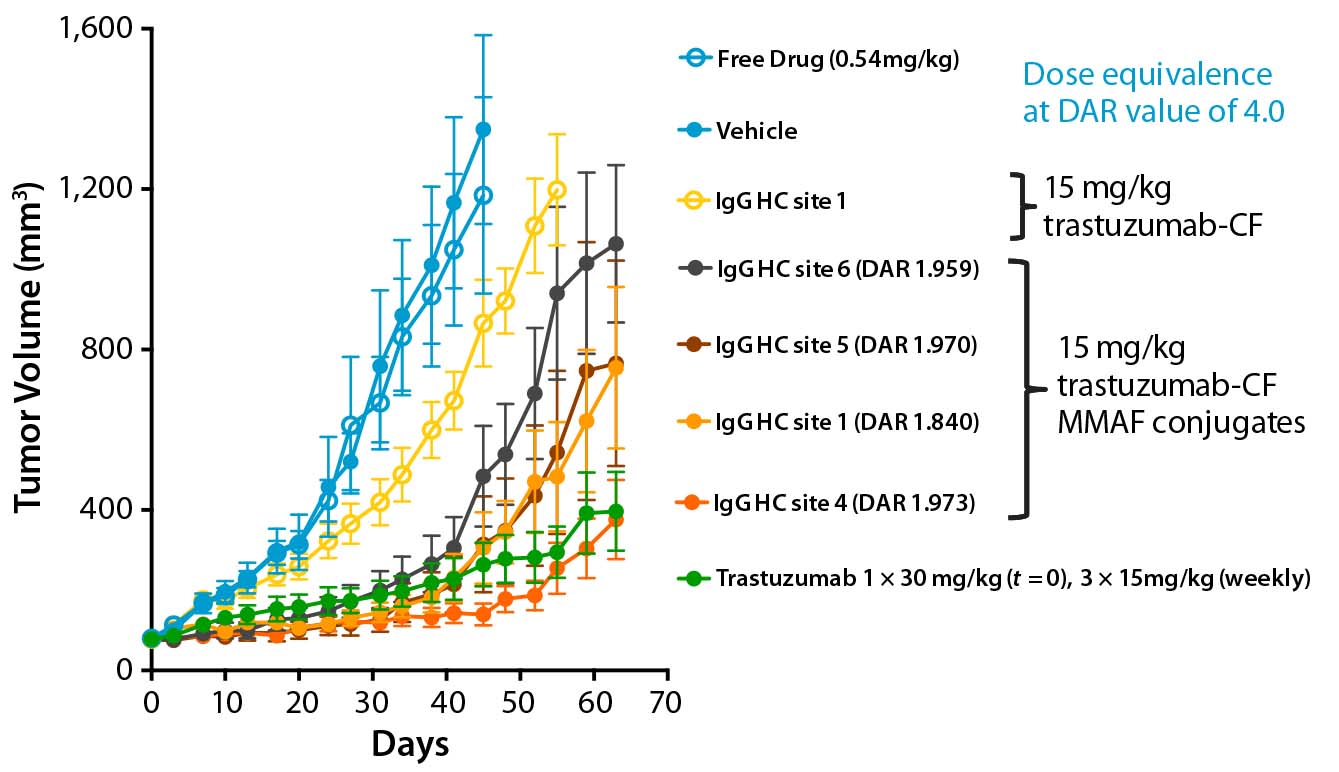
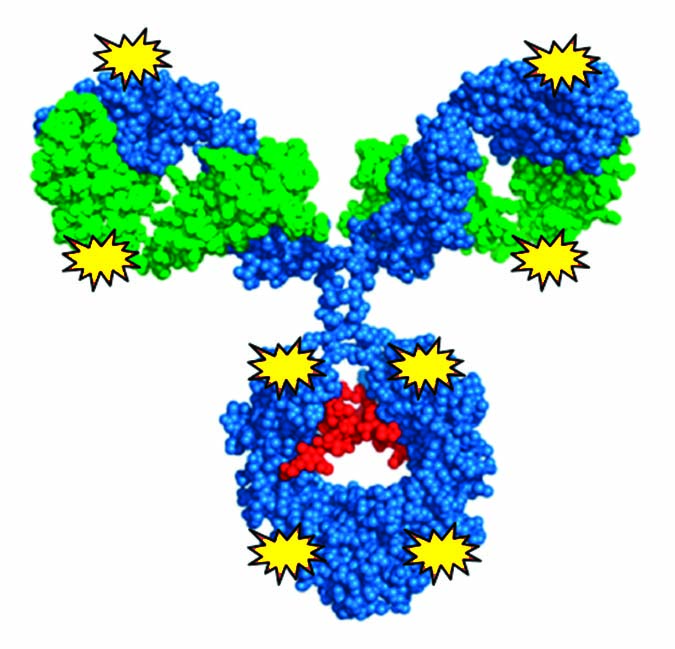
Figure 1: ADC efficacy is conjugation-site dependent.
Using a scalable biochemical protein synthesis platform, our company constructed libraries of nnAA-containing conjugation-site variants of antibodies to find the best available sites for conjugation (6). Clearly, positioning of the conjugated linker–warhead matters (Figure 1). Some positional variants can outperform both other homogeneous variants and heterogeneous ADCs based on the same linker–warhead and antibody combination.
Selecting the Optimal Conjugation Site for Best-in-Class ADCs: Homogeneity and conjugation site predictability play a significant role in efficacy and pharmacokinetics of an ADC and influence its stability, conjugation efficacy, immunogenicity, antigen-binding, and internalization characteristics. It is important to determine the precise conjugation site(s) for each particular antibody, linker, and warhead combination and select the best ADC species for an optimal efficacy and safety profile. There are 300–400 potential positions would have to be expressed and characterized to identify the best conjugation site. Expressing so many individual proteins is a challenge for even state-of-the-art cell-based expression systems. However, scalable biochemical protein synthesis platforms based on Stanford professor James R. Swartz’s patented open cell-free synthesis (OCFS) technology overcome such limitations.
The Xpress CF and Xpress CF+ platforms involve an extract that contains cellular components required to produce proteins. The extract includes all necessary biochemical components for energy production, transcription, and translation and can be used to support cell-free biochemical protein synthesis with addition of the specific DNA sequence for a desired protein. And establishing a cell line for each variant becomes unnecessary. A biochemical protein production reaction can be carried out at very small volumes (in microplates), which enables rapid production and characterization of variants by the hundreds. Note that the reaction can be scaled up quickly and easily for production of single proteins at gram-per-liter yields in 8–10 hours at any scale (7).
Another specific attribute of scalable biochemical protein synthesis is that the platform is amenable to efficient incorporation of novel nnAAs, such as para-azidomethyl-l-phenylalanine (pAMF or pAz-Me- Phe). Efficiency of the conjugation reaction is a key concern when manufacturing ADCs. Use of pAMF enables more efficient conjugation kinetics than other nnAAs such as pAz-Phe (“click” chemistry) or pAc- Phe (oxide ligation chemistry), which are preferred for intact cell-expression systems because of their plasma membrane permeability. The absence of cells in biochemical protein production allows for use of more reactive but less cell-membrane– permeant nnAAs to enhance manufacturing feasibility.
Our company has demonstrated that site-specific incorporation of pAMF enabled complete conjugation of a DBCO-PEG-monomethyl auristatin (DBCO-PEG-MMAF) drug to the Her2-binding IgG trastuzumab using azide−alkyne cycloaddition (SPAAC) copper-free click chemistry within two to four hours at room temperature, with very little molar excess of linker–warhead (8). A novel variant of the established Methanococcus jannaschii tyrosyl tRNA synthetase (TyrRS) with a high activity and specificity toward optimized pAMF was discovered by using the scalable Xpress CF and Xpress CF+ biochemical protein synthesis platforms to screen a library of aminoacyl tRNA synthetase (aaRS) variants. It enables efficient production and manufacturing of pAMF-containing ADCs.
Manufacturing efficiency also depends on how efficiently an nnAA-containing antibody can be produced. It should be noted that biochemical expression systems are devoid of RF-1, a release factor that terminates translation by recognizing the amber stop codon. In the presence of RF-1, titers of nnAA-containing antibodies are reduced to 30–40% of the natural antibody titer. Such inefficiency is unavoidable in cell-based expression systems because RF-1 is critical for cell growth.
What’s to Come?
Today, ADC developers are facing two challenges:
- how to increase clinical impact for tumors that display heterogeneous expression of a target antigen
- how to improve outcomes for those ADCs that show promising early efficacy before tumors develop resistance.
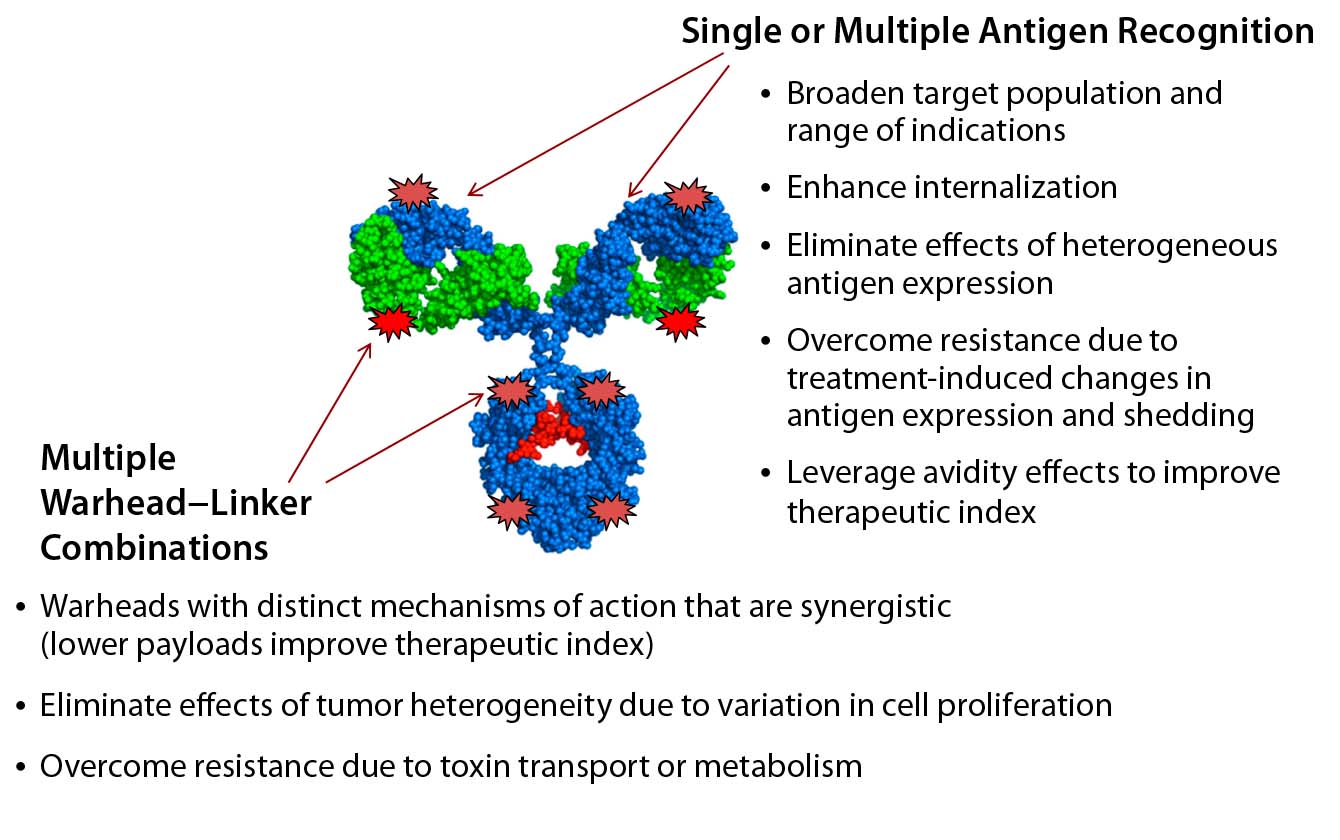
Figure 2: The next generation of ADCs will be homogeneous and multifunctional.
In part, both challenges could be addressed by generating new ADC formats that recognize multiple tumor antigens — e.g., drug conjugates based on bispecific antigen targeting — or by delivering multiple and complementary warhead types (Figure 2).
Recently, our company showed that the biochemical protein expression platform also allows for generation and manufacture of dual-warhead ADCs (9). Such biologics include two different cytotoxic agents attached to a targeting antibody, potentially addressing the challenge of drug resistance in cancer therapies. Combining mechanistically synergistic warheads would be important for decreasing the load of either warhead for patients while maintaining superior efficacy against their tumors. That could well provide greater clinical impact from an efficacy and safety perspective. Our team precisely incorporated two distinct nnAAs with mutually orthogonal chemistry (azide-based and tetrazine-based) onto one antibody and showed that a single conjugation reaction could generate homogeneous dual-warhead ADCs (Figure 3).
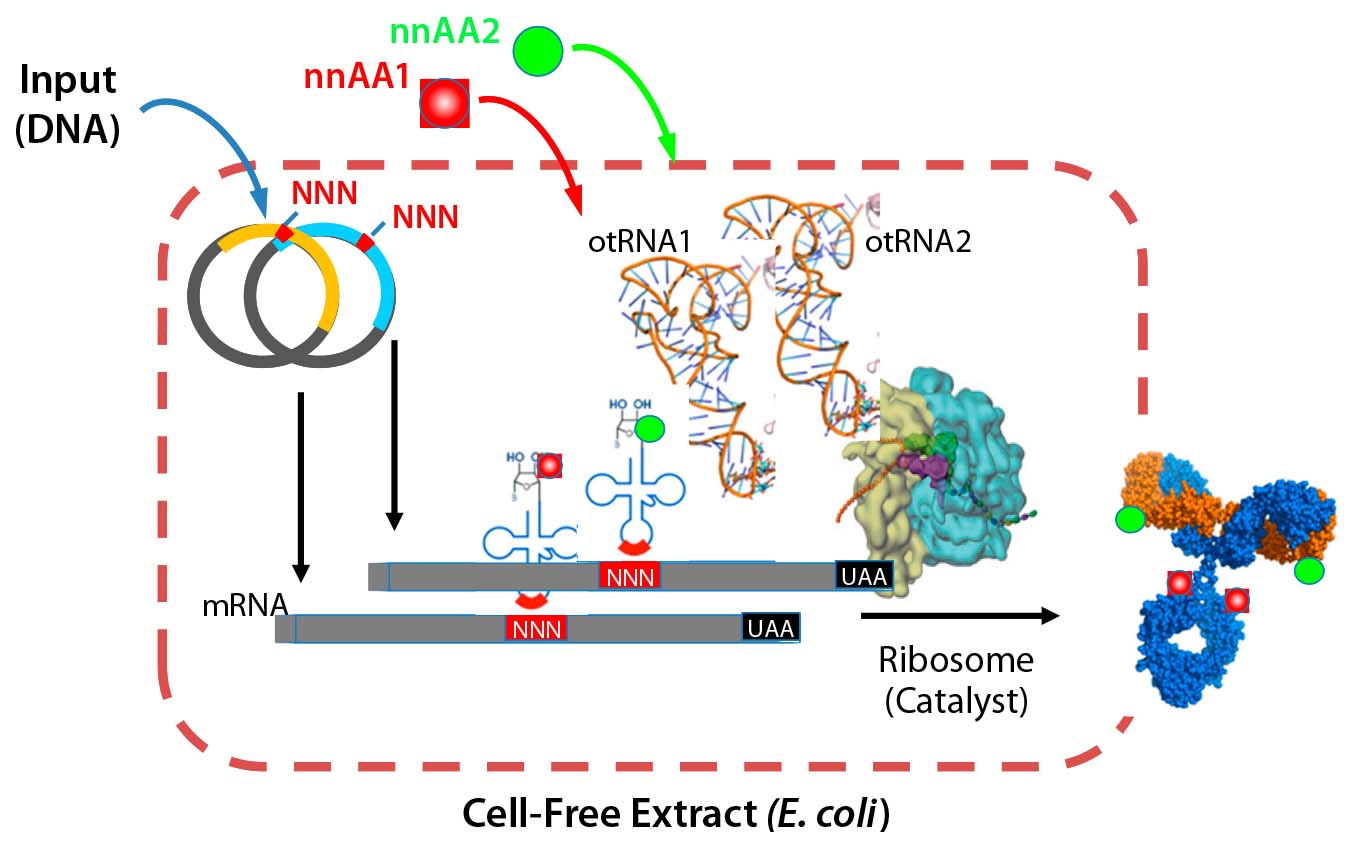
Figure 3: Xpress CF and Xpress CF+ scalable biochemical protein synthesis platforms enable
generation and manufacturing of biotherapeutics including proteins containing one or more nnAAs for site-specific conjugation in homogeneous ADCs.
Creating best-in-class ADCs by producing homogeneous molecules has tremendous potential to optimize clinical results by improving efficacy and product safety. Homogeneity in itself is insufficient. Systematic design of these complex molecules will require real drug-discovery–based reiterative design and development of structure–activity relationships to guide design of optimized molecules. Biochemical protein expression may be one such solution.
References
1 Antibody–Drug Conjugates Market, 2013–2023. Roots Analysis: London, UK, 22 July 2013; www.rootsanalysis.com/reports/ view_document?report_id=21.
2 Behrens CR, Liu B. Methods for Site- Specific Drug Conjugation to Antibodies. MAbs 6(1) 2014: 46–53.
3 Junutula JR, et al. Site-Specific Conjugation of a Cytotoxic Drug to an Antibody Improves the Therapeutic Index. Nature Biotechnol. 26(8) 2008: 925–932.
4 Wang L, et al. Expanding the Genetic Code of Escherichia coli. Science 292 (5516) 2001: 498–500.
5 Jackson D, et al. In Vitro and In Vivo Evaluation of Cysteine and Site-Specific Conjugated Herceptin Antibody–Drug Conjugates. PLoS One 14 January 2014; www.plosone.org/article/ info%3Adoi%2F10.1371%2Fjournal.pone.0083865.
6 Hallam TJ, Smider VV. Unnatural Amino Acids in Novel Antibody Conjugates. Future Med. Chem. 6(11) 2014: 1309–1324.
7 Zawada JF, et al. Microscale to Manufacturing Scale-Up of Cell-Free Cytokine Production: A New Approach for Shortening Protein Production Development Timelines. Biotechnol. Bioeng. 108(7) 2011: 1570–1578.
8 Zimmerman ES, et al. Production of Site-Specific Antibody–Drug Conjugates Using Optimized Non-Natural Amino Acids in a Cell-Free Expression System. Bioconjug. Chem. 25, 2014: 351–361.
9 Hallam TJ. Tumor Heterogeneity and Resistance: Production of Homogeneous Bispecific ADCs with Combination Warheads. ADC Summit 19–20 May 2014: London, UK.
Trevor Hallam, PhD, is chief scientific officer; Jeremy Bender, PhD, is chief business officer; and Bill Newell is chief executive officer of Sutro Biopharma, 310 Utah Avenue, Suite 150, South San Francisco, CA 94080; 1-650-392-8412, fax 1-650-872-8924; www.sutrobio.com.