According to the late Norman Cousins, “Wisdom consists of the anticipation of consequences.” When it comes to regulatory inspections, those consequences can be severe. However, the consequences of a problem anticipated can be prevented — given effective action to remediate the issue. In two previous articles (1, 2), I discussed the whys and hows of using the US Food and Drug Administration's (FDA's) notices of deficiency, FDA warning letters, and other information about inspection results to create an effective system to spot and rectify your own compliance issues before they become adverse inspectional outcomes. Here, I provide an analysis of more than 40 FDA warning letters to identify inspectional trends for cellular and tissue-based products.

The Regulatory Landscape
The FDA's attempt to regulate human cells, tissues, and cellular and tissue-based products (HCT/Ps) emerged from its historic fragmentation in 1997 with the proposal of a new approach and an accompanying public meeting, first announced in the Federal Register(3). The agency stated
The Food and Drug Administration (FDA) is announcing the availability of a document entitled, Proposed Approach to Regulation of Cellular and Tissue-Based Products. In addition, FDA is announcing a public meeting to solicit information and views from the interested public on the agency's proposed regulatory approach for such products. Those actions are taken in response to the administration's Reinventing Government initiative which seeks to streamline regulatory requirements to ease the burden on regulated industry, while providing adequate protection to the public health.
Subsequently, to implement this proposed new approach, the agency published three final rules and two interim final rules. The final rules were Human Cells, Tissues, and Cellular and Tissue-Based Products: Establishment Registration and Listing (2001), Eligibility Determination for Donors of Human Cells, Tissues, and Cellular and Tissue-Based Products (2004), and Current Good Tissue Practice for Human Cell, Tissue, and Cellular and Tissue-Based Product Establishments, Inspection and Enforcement (2004) (4,5,6). The latter final rule, codified as 21 CFR 1271 (Title 21 of the Code of Federal Regulations, Part 1271) is known as the Current Good Tissue Practices (CGTPs). The interim final rules were 2004's Human Cells, Tissues, and Cellular and Tissue-Based Products; Establishment Registration and Listing and 2005's Human Cells, Tissues, and Cellular and Tissue-Based Products; Donor Screening and Testing, and Related Labeling” (7,–8). In August 2007, FDA further clarified regulatory expectations in an industry guidance document on HCT/P compliance (9). Underscoring the importance of the subject, that guidance was released “for immediate implementation,” rather than being first released in draft form, as are the majority of guidance documents.
Despite the dogs of controversy barking about FDA's jurisdiction over stem cell therapies and the precise meaning of “minimally manipulated” (10), the regulatory caravan moves on. This includes, among other things, regulatory inspections of HCT/P companies for compliance with current good manufacturing practice (CGMP) as contained in 21 CFR Parts 211 and 820 (the device GMPs called QSRs — quality system regulations), the human tissue intended for transplantation regulations contained in 21 CFR Part 1270, and current good tissue practice (CGTP) in 21 CFR Part 1271. Additional detail about the HCT/P regulatory environment are provided elsewhere (11, 12), and inspections and 21 CFR regulations are listed on www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm.
Inspection Results and Inspection Trends
To look for trends in inspections related to the production of cellular and tissue-based products, I analyzed 41 FDA warning letters sent to HCT/P companies after adverse inspections. Those letters spanned the years 2000 through 2013. Represented companies in the warning letters include those in cell and tissue processing, banking, and therapy as well as those for human reproductive cells and tissues. The latter often have the purpose of in-vitro fertilization (IVF).
Inspection teams often know that a deficiency at one company will often be found at others, so they tend to look for what teams have found in previous inspections. A numerical analysis of past inspections results in the discovery of inspection trends for future inspections (Tables 1 A–C and 2). Although only warning letters are analyzed herein, HCT/P 483s are discussed in other sources (13, 14).
Table 1A
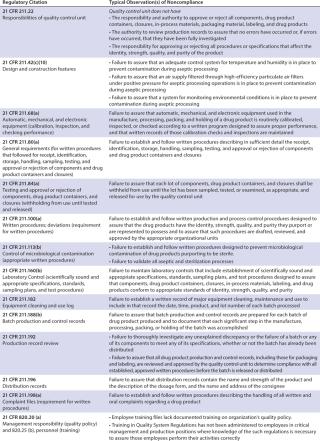
Table 1B
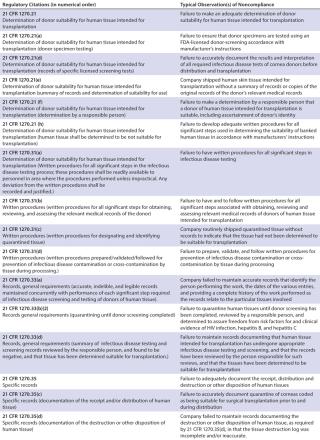
Table 1C
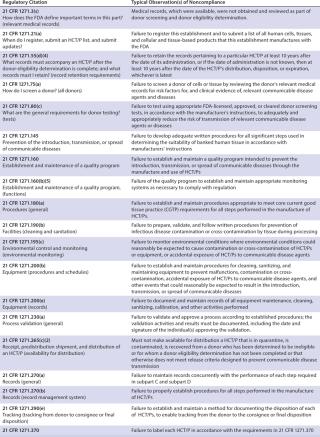
Table 2
The warning letters over this 14-year period were split almost evenly among companies that could be classified as cell/tissue processing/banking/therapy companies and those whose primary purpose is restricted to IVF, with 21 letters going to the former and 20 going to the latter. A signal difference, however, is that only 21 CFR Part 1271 (CGTPs) was named in the warning letters to the human reproductive cell/tissue (IVF) companies, whereas warning letters to other cell/tissue processing/banking/therapy companies cited a variety of regulations. Those regulations include not only CGTPs, but also 21 CFR Part 1270 (human rissue intended for rransplantation), 21 CFR Part 211 (CGMPs), and even a citation from 21 CFR 820 (medical device QSRs).
The warning letters were not spread out evenly over those years, as might be expected by growth of both the number of new companies and HCT/P-related activities at existing companies. A little more than one-third (15, 37%) of the letters were issued in the seven-year period from 2000–2006, and almost two-thirds (26, 63%) were issued during the seven-year period 2007–2013. The peak thus far came in 2012, with seven warning letters issued, but 2013 saw a decline, with four letters sent. In all, over 60 regulations (counting subparts) were cited in those letters, with 22% coming from the CGMPs (including the one QSR citation), 55% coming from the CGTPs, and the remaining 23% coming from 21 CFR Part 1270.
Overall, the most cited regulation was 21 CFR 1271.85 (15% of all citations). It deals with donor screening, as do 1271.75 (11% of all citations) and 1271.80 and 1270.31 (each 10%). 21 CFR 1271.47, which also regulates donor screening, made up 6% of all citations. The fifth most cited regulations (each 5% of total), were 21 CFR 1270.21 and 1271.55, whose subjects are, respectively, donor screening and donor eligibility records.
In all, donor screening and documentation issues made up slightly over 60% of all warning letter citations when categorized by 21 CFR part/subpart number. Other important issues were 21 CFR 1270.33, general documentation and records requirements (4%); 1270.35, documentation of receipt, quarantine, distribution, and destruction/disposition of tissues (3%); and 1271.150, general requirements of CGTP (3%). The tables list other, less-cited regulations.
Considering the topics and subjects most cited — rather than citations by 21 CFR part number — donor screening/testing/documentation issues accounted for almost half (49%) of the citations when they were analyzed in this way. The next most frequent topic cited was the ever-present documentation/recordkeeping (other than for donors) at 21%, followed by issues with a deficient quality unit (9%), quarantine/withholding from use issues (4%), deficient processing operations (also 4%), and inadequate process validation (3%). Labeling issues, inadequate environmental monitoring, and deficient equipment calibration/maintenance/performance checking each accounted for 2% of the adverse inspections results, and deficient facilities design/construction, failure/deviation/discrepancy investigations, training inadequacies, cleaning and sanitation deficiencies, and contract service provider issues each accounted for 1% of the citations. Other issues, undoubtedly important but accounting for less than 1% each of the citations were deficient product testing, inadequate handling of complaints, and failure to register the establishment.
Taking Precautions
The analysis here clearly shows that much compliance attention should be paid to the systems in place for donor screening and documentation of that screening because they were the overwhelmingly most-cited issues in regenerative medicine warning letters so far. In addition, documentation and recordkeeping issues other than those having to do with donors loom large as does having an adequate quality unit and quality systems. I hope this analysis will help your company be ready when the next inspection team comes knocking. Readers may be able to tease still more knowledge from the data I have given. Happy mining!
Author Details
BPI Editorial Advisory Board member Tom Pritchett, PhD, is the publisher of the biopharmaceutical/biologics industry newsletter BioQuality and an industry consultant and trainer;
REFERENCES